Fig. 8.1
Calcitonin stain showing C-cell hyperplasia (Courtesy of Dr Moosa Khalil)
MTCs are most frequently located posteriorly at the junction between the upper-third and lower two-thirds of the lateral lobes of the thyroid where the C-cells are concentrated. They are usually firm, white or grey, and well defined but unencapsulated (Fig. 8.2). Cells can be variable in shape and mildly pleomorphic but nuclei are characteristic of other neuroendocrine tumours, with round-oval shape with a fine stippled ‘salt and pepper’ nuclear chromatin (Fig. 8.3a). Calcification and surrounding amyloid (composed of full-length calcitonin) might be present (Fig. 8.3b) [11, 12].
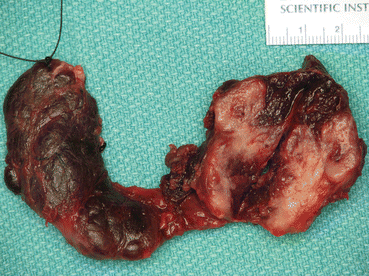
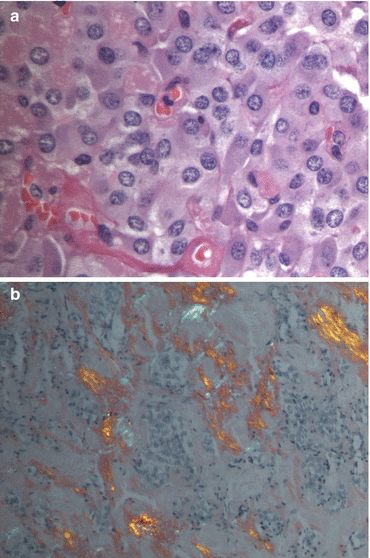
Fig. 8.2
Resected thyroid showing medullary thyroid cancer in the left lobe (Courtesy of Dr Moosa Khalil)
Fig. 8.3
(a) Haematoxylin and eosin stain of medullary thyroid cancer. (b) Congo red stain under polarized light showing amyloid in medullary thyroid cancer (Courtesy of Dr Moosa Khalil)
Genetic Testing
The 21 exon RET proto-oncogene on chromosome 10q11.2 was first described in 1985 [13]. The RET proto-oncogene encodes the RET (rearranged during transfection) protein, a receptor tyrosine kinase (RTK) that is expressed in neural crest-origin derivatives [14]. There are 17–20 classes of 58 RTKs that also include vascular endothelial growth factor (VEGF) and endothelial-derived growth factor receptor (EGFR). RTKs consist of a ligand-binding extracellular portion, a cysteine-rich region necessary for receptor dimerization, a transmembrane domain, and an intracellular portion consisting of two tyrosine kinase (TK) regions that activate intracellular signal transduction pathways (Fig. 8.4) [15]. Activation triggers autophosphorylation of the extracellular tyrosine residues that serve as docking sites for adaptor proteins, which coordinate intracellular signal transduction pathways that are important in regulating cell growth [1–3, 14].
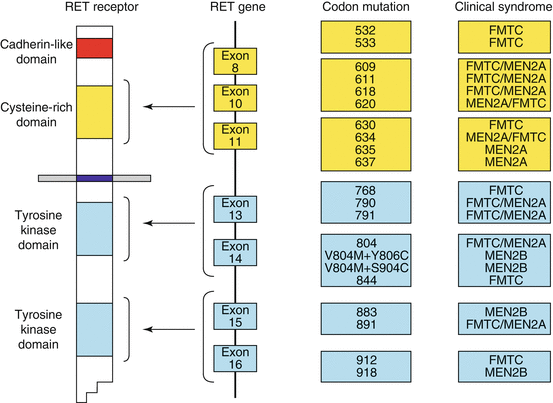
Fig. 8.4
Diagram of the RET receptor, gene and corresponding codon mutations and their associated clinical syndromes (Reproduced with permission from Hubner and Houlston [15])
Germline point mutations in the RET proto-oncogene can be identified in 95 % of cases of MEN2 (multiple endocrine neoplasia type 2), which suggests that undiscovered additional loci may exist (Fig. 8.4). In sporadic MTC, germline RET mutations are uncommon (1.5–24 %) but somatic RET mutations occur in 25–50 % of cases [4, 5, 16–18].
Hereditary MTC
Hereditary forms of MTC account for ~30 % of MTC. MEN2A and 2B are inherited in an autosomal dominant fashion with complete penetrance for MTC. MTC can present at any age but is most common in the second to fourth decades [6, 19]. It is usually the first presentation of the syndrome and is often multicentric and bilateral. Genetic testing may also reveal family members positive for germline mutations who can be considered for prophylactic surgery before the development of MTC.
MEN2A (Sipple syndrome) is the most common variant of MEN2, occurring in 80–90 % of familial MTC. The most common codon mutation is 634 (80 %). The MEN2A consists of MTC (>90 %), pheochromocytoma (50 %) and primary hyperparathyroidism (l5–30 %). Index cases of MTC often present with a thyroid or neck mass between the ages of 15 and 20 years, although it has been observed in children as young as 5 years of age [7, 20]. Variations of MEN2A also occur uncommonly with RET codon 634 mutation with cutaneous lichen amyloidosis (upper back cutaneous pruritic lesion) or with Hirschprung disease [8, 21, 22].
Familial MTC (FMTC) was described initially in 1986 in two families who were developed a more indolent form of MTC at a later age (~43 years old) with no other clinical manifestations of the MEN2 syndrome [6, 9, 10, 23]. The penetrance of MTC is lower than that of MEN2A and occurs at an older age. Definition of FMTC varies from greater than four members in family with MTC without pheochromocytoma or hyperparathyroidism to more stringent guidelines of more than 10 carriers with multiple carriers, or affected members over the age of 50 years [11, 12, 19, 24]. The stricter criteria aim to avoid mislabelling late occurring MEN2A as FMTC and potentially miss a pheochromocytoma [13, 19]. This may also account for the difference in the incidence of FMTC and MEN2A in the literature. In a large series, 75 % of FMTC presented as apparently sporadic cases [14, 25]. For this reason, FMTC could be considered as a variant of MEN2A with decreased penetrance for pheochromocytoma and hyperparathyroidism, rather than as its own entity.
MEN2B occurs in 5–20 % of MEN2 patients. Features include: (i) MTC; (ii) pheochromocytoma; (iii) marfanoid habitus (without Marfan syndrome); (iv) pes cavus; (v) pectus excavatum; (vi) hypotonia; (vii) proximal muscle weakness; (viii) mucosal neuromas on the lips, anterolateral surface of tongue and conjunctiva; (ix) medullated corneal nerves; and (x) intestinal ganglioneuromatosis [24]. The most common mutation is codon 918 in 95 % of patients with MEN2B. Approximately 50 % of MEN2B patients have de novo germline RET mutations, usually of paternal origin [26]. In MEN2B, both MTC and pheochromocytoma are more aggressive variants that occur at an earlier age compared with MEN2A.
Pheochromocytoma in MEN2 are usually bilateral (≤78 %), rarely metastasize (<5 %), and half are asymptomatic. Annual surveillance with 24-h urine metanephrines or plasma-free metanephrines is recommended from the age of 8 years for MEN2B and codons 630 and 634, and by the age of 20 years for other MEN2A mutations. The difficulty with surveillance lies in differentiating pheochromocytoma from its precursor—adrenal medullary hyperplasia—when a patient has elevated biochemical tests but an absence of localizing imaging [20].
Before the era of genetic testing, screening those at high-risk for familial MTC was with yearly calcitonin and pentagastrin-stimulated calcitonin levels, 24-h urinary metanephrines, serum calcium, and parathyroid hormone (PTH) measurement in 6–35-year-olds. Currently, all first-degree relatives of known germline RET mutation carriers should be offered genetic testing before the age of recommended prophylactic surgery [27]. DNA is specifically examined for the exons commonly involved with MEN2, eliminating the need for yearly screening of all family members. The guidelines recommend that children who are positive for RET mutation undergo prophylactic thyroidectomy. Many ethical considerations surround genetic testing and prophylactic surgery at such a young age, but the accuracy of RET testing is high (95 % for MEN2 and 88 % for FMTC), penetrance of MTC is 100 %, and surgery is the only chance for cure [28]. The location of the RET codon mutation can predict biological behaviour, and guidelines stratifying codon types into four risk levels have been established to direct the timing of prophylactic surgery (Table 8.1).
Table 8.1
Prophylactic surgery by codon mutation
ATA risk level | Codons | Subtype | RET testing | Age for ultrasound | Age for first serum calcitonin | Age for surgery |
|---|---|---|---|---|---|---|
D (highest risk) | 883 918 | MEN2B | ASAP, within 1st year of life | ASAP, within 1st year of life | 6 months if surgery not already performed | ASAP, within 1st year of life |
C | 634 | MEN2A | <3–5 years | >3–5 years | >3–5 years | Before 5 years lymph node dissection controversial |
B | 609 611 618 620 630 | MEN2A FMTC | <3–5 years | >3–5 years | >3–5 years | Total thyroidectomy 5–10 years; some suggest by 5 years |
A (lower risk) | 768 790 790 804 891 | MEN2A FMTC | <3–5 years | >3–5 years | >3–5 years | Total thyroidectomy 5–10 years |
Prophylactic surgery for paediatric MEN2 patients (ATA-A − D) with no evidence of metastases is total thyroidectomy. MEN2B patients treated after the age of 1 year should also undergo a prophylactic central compartment dissection. Preoperatively, elevated serum calcitonin of >40 pg/ml or MTC size of ≥5 mm indicate the need for investigation for distant metastases, although the accuracy of calcitonin to predict metastases in very young patients is unknown. Therapeutic dissection of positive lymph node compartments should be undertaken.
Primary hyperparathyroidism in MEN2A can be solitary or multiple and is more likely to coexist if codon 634 mutation is present. Normal parathyroid glands should be tagged and left in situ, if possible. In MEN2A, abnormal parathyroid glands seen at MTC surgery should be treated with a parathyroidectomy and autograft into the forearm, as if the patient has mild hyperparathyroidism, even if the patient is normocalcaemic [19]. This will also protect the parathyroid function in the event of reoperative surgery for either recurrent MTC or hyperparathyroidism. For MEN2B or FMTC, devascularized parathyroids should be autotransplanted. Some surgeons leave the parathyroid glands in situ when treating hereditary MTCs if the patient is <8 years old, as they have found the risk of nodal metastases to be is low [29].
Newly Detected Cases of MTC
MTC may be detected during routine screening when calcitonin is used to monitor thyroid nodular disease, a practice that has caused much debate. Large European series have found a 0.4–0.6 % incidence of elevated calcitonin in thyroid nodules with fine-needle aspiration (FNA) that is not diagnostic of MTC [30, 31]. These patients were appropriately treated for MTC at an earlier stage with subsequent postoperative calcitonin normalization [30, 31]. However, screening calcitonin, especially when only mildly elevated, might lead to surgery for CCH or even benign disease [32]. Although calcitonin has good sensitivity for diagnosing MTC, confirmation with a pentagastrin stimulation test has been suggested [33]. In North America where pentagastrin is unavailable, interpretation of mildly elevated calcitonin levels is difficult [34]. Calcium-stimulated calcitonin has been shown in relatively smaller studies to be well tolerated, and stimulated-calcitonin levels distinguishing normal, CCH and MTC have been identified [35]. Screening for calcitonin appears to be cost-effective in evaluation models in both Europe and USA [36, 37]. Despite the earlier recommendation for screening calcitonin, the most recent European guidelines are less enthusiastic and instead suggest that it might be a useful nodular disease while the National Comprehensive Cancer Network (NCCN) guidelines have remained equivocal [27, 38].
Sporadic MTC accounts for 70 % of MTCs and usually presents in the fifth/sixth decade of life as a palpable thyroid mass or lymph node (LN), anterior neck pain, compressive symptoms of hoarseness, dysphagia or dyspnoea or hormonal symptoms, such as flushing or diarrhoea. [39] A thorough history should be taken with emphasis on the possibility of previously undetected familial disease. At presentation, LN involvement occurs in 35–50 % of patients and distant metastases in 10–15 %, most commonly in the lungs, mediastinum, liver, bone and less commonly to brain and skin.
Investigations to assess locoregional disease include high-resolution ultrasound of the thyroid, central and bilateral lateral LN compartments, and superior mediastinum. FNA of suspicious nodules or LNs should be undertaken. The accuracy of FNA for MTC is lower than that in differentiated thyroid cancer, with only 62 % sensitivity [40]. As an adjunct, following the FNA, the needle can be washed in 1 ml of normal saline and sent for calcitonin testing (calcitonin washout). This can detect MTC in thyroid nodules and metastatic LNs with almost 100 % sensitivity and specificity [40].
Serum tumour markers, in particular calcitonin, aid in staging, follow-up and prognosis. Calcitonin levels correlate with tumour burden, LN metastases (likely if calcitonin levels between 10–40 pg/ml), and distant metastases (highly likely if >1,000 pg/ml) [41, 42]. Preoperative serum carcino-embryonic antigen (CEA) might also correlate with tumour burden [43]. Serum chromogranin A, often used to detect neuroendocrine tumours, has limited utility in MTC, as it is elevated only in advanced disease [44].
All patients with MTC, CCH or MEN2 should be offered genetic testing for germline RET mutations of known MEN2-related exons. New patients presenting with apparently sporadic MTC have germline RET mutations in 4–6.5 % and 41.1 % of their tested relatives are gene carriers [45, 46]. Routine testing for somatic RET mutations is not currently recommended by the NCCN guidelines, although their presence may indicate a more advanced stage at diagnosis with worse prognosis for persistent disease and death, although this has not been correlated in all studies [45].
Preoperatively, pheochromocytoma should be excluded with either negative RET testing, 24-h urinary metanephrines, or plasma meta-nephrines and adrenal imaging with CT or MRI. If pheochromocytoma is diagnosed, it must be treated with appropriate preoperative alpha-blockade before MTC to avoid the complication of an intraoperative adrenergic crisis. Serum calcium and PTH should be measured before surgery to exclude primary hyperparathyroidism and aid in surgical planning.
Management of Locoregional Disease
Surgery for disease confined to the thyroid (T1-3, N0, M0) is total thyroidectomy and prophylactic central (level VI) LN dissection. In unilateral palpable MTC, central LNs are involved in 81 % [47]. The central compartment has been defined as the carotid sheaths laterally, the hyoid superiorly, the innominate artery or sternal notch inferiorly, and the superficial and deep layers of the deep cervical fascia anteriorly and posteriorly [48].
Lateral neck dissection (therapeutic) is recommended if there is suspicion of lateral LN disease either clinically or radiologically in those LN basins. For those with palpable LN at presentation, the rate of LN involvement has been found to be high, with 100 % central compartment, 93 % ipsilateral lateral, 45 % contralateral, and 52 % mediastinal involvement [49]. The importance of preoperative high-resolution ultrasound is emphasized, as it is difficult even for experienced surgeons to reliably identify LN metastases intraoperatively by palpation and inspection (sensitivity 64 %, specificity 71 %) [47].
The role of prophylactic lateral LN dissection is controversial. The recent ATA guidelines have recognized, with value of ultrasound to predict disease in the lateral compartments and suggest FNA of suspicious nodes and subsequent compartment dissection, only for proven positive disease [27]. However, proponents of prophylactic lateral LN dissection argue that in the absence of effective adjuvant therapy for a disease with high rates of occult LN metastases, surgery should be thorough. In unilobar MTC, ipsilateral lateral LN metastases occur in 57–81 % and contralateral disease in 28–44 % [47, 50] The risk of LN metastases in multifocal disease is at least doubled compared with unifocal disease [51]. Although the size of MTC has been found to be a predictor of LN involvement, it is inconsistent [50, 52].
Involved central compartment LNs may predict the presence and number of ipsilateral and to a lesser extent contralateral lateral LN metastases [53]. Contralateral LNs are usually only involved in addition to positive central and ipsilateral lateral LNs [53]. LN status may also be able to predict an undetectable postoperative calcitonin with 95 % undetectable without LN metastases, and 32 % with LN metastases [50]. Bilateral lateral LN dissection may be considered in the presence of extensive ipsilateral lateral LN disease [54]. However, despite extensive surgery with its concomitant risks, even N0 patients may not have calcitonin normalized in 38 % of them, which makes the benefit of prophylactic lateral LN dissection questionable [42]. An alternative strategy might be to initially defer the prophylactic lateral LN dissection until postoperative calcitonin levels are indicative of surgery.
Mediastinal metastatic LN may indicate systemic disease and confer poor prognosis; thus suspicious upper mediastinal nodes should be removed via the cervical incision, if possible [55, 56]. Compartment-orientated dissection of mediastinal LN via a trans-sternal approach has been performed previously by some groups, but is now considered only in exceptional circumstances [56].
In the presence of extensive local disease or distant metastases, surgery might be less aggressive and tailored to a more palliative approach aiming to minimize complications and to control symptoms, such as pain or airway compromise. Diagnostic laparoscopy has been used to detect liver metastases that are too small to be seen on cross-sectional imaging [57]. Surgery should be planned by bearing in mind staging and predicted survival with their burden of metastatic disease. Systemic therapies or clinical trials may be considered.
Staging and Prognosis
Staging is important to plan the extent of surgery, as a less aggressive approach may be taken in the setting of locally advanced or advanced metastatic disease. Investigations for distant metastases are CT of the neck and mediastinum with additional CT of the abdomen and pelvis if calcitonin is >400 pg/ml. The AJCC TNM staging is listed in Tables 8.2 and 8.3.
 Management of the Lateral Neck in Well-Differentiated Thyroid Cancer
Anaplastic Thyroid Cancer: Current Concepts
The Significance of Cervical Lymph Nodes in Well-Differentiated Thyroid Cancer
Management of the Lateral Neck in Well-Differentiated Thyroid Cancer
Anaplastic Thyroid Cancer: Current Concepts
The Significance of Cervical Lymph Nodes in Well-Differentiated Thyroid Cancer
 External Beam Radiotherapy for Thyroid Cancer
External Beam Radiotherapy for Thyroid Cancer
 Management of Distant Metastases in Differentiated Thyroid Cancer
Management of Distant Metastases in Differentiated Thyroid Cancer
 Management of the Central Compartment in Well-Differentiated Thyroid Carcinoma
Management of the Central Compartment in Well-Differentiated Thyroid Carcinoma




Table 8.2
American Joint Committee on Cancer—tumour, node, metastasis (TNM) staging for thyroid cancer [58]
Primary tumour (T) | |
Tx | Primary tumour cannot be assessed |
T0 | No evidence of primary tumour |
T1 | Tumour ≤2 cm in greatest dimension, limited to the thyroid |
T1a | Tumour ≤1 cm, limited to the thyroid |
T1b | Tumour >1 cm but not >2 cm in greatest dimension, limited to the thyroid |
T2 | Tumour >2 cm but not >4 cm in greatest dimension, limited to the thyroid |
T3 | Tumour >4 cm in greatest dimension, limited to the thyroid or any tumour with minimal extrathyroid extension (e.g., extension to sternothyroid muscle or perithyroid soft tissues) |
T4a | Moderately advanced disease |
Tumour of any size extending beyond the thyroid capsule to invade subcutaneous soft tissues, larynx, trachea, oesophagus, or recurrent laryngeal nerve | |
T4b | Very advanced disease |
Tumour invades prevertebral fascia or encases the carotid artery or mediastinal vessels | |
Regional lymph nodes (N) | |
Nx | Regional lymph nodes cannot be assessed |
N0 | No regional lymph node metastasis |
N1 | Regional lymph node metastasis |
N1a | Metastasis to level VI (pretracheal, paratracheal and prelaryngeal/Delphian lymph nodes) |
N1b | Metastasis to unilateral, bilateral or contralateral cervical (levels I, II, III, IV or V) or retropharyngeal or superior mediastinal lymph nodes (level VII) |
Distant metastasis (M) | |
M0 | No distant metastasis |
M1 | Distant metastasis |
Stage group | T stage | N stage | M stage | 5-year survival (%) |
|---|---|---|---|---|
Stage I | T1 | N0 | M0 | 98–100 |
Stage II | T2 | N0 | M0 | 93–100 |
Stage III | T3 | N0 | M0 | 66–73 |
T1 | N1a | M0 | ||
T2 | N1a | M0 | ||
T3 | N1a | M0 | ||
Stage IV | 21–48 | |||
Stage IVA | TIa | N0 | M0 | |
TIa | N1a | M0 | ||
T1 | N1b | M0 | ||
T2 | N1b | M0 | ||
T3 | N1b | M0 | ||
T4a | N1b | M0 | ||
Stage IVB | T4b | Any N | M0 | |
Stage IVC | Any T | Any N | M1 |
Reproduced with permission from Edge et al. [58]:89
Related posts:
Management of the Lateral Neck in Well-Differentiated Thyroid Cancer
Anaplastic Thyroid Cancer: Current Concepts
The Significance of Cervical Lymph Nodes in Well-Differentiated Thyroid Cancer
External Beam Radiotherapy for Thyroid Cancer
Management of Distant Metastases in Differentiated Thyroid Cancer
Management of the Central Compartment in Well-Differentiated Thyroid Carcinoma
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
