Fig. 1.1
Translation initiation and common mechanisms of regulation. The steps of translation initiation are outlined in (a). The ternary complex is comprised of eIFs 1, 1A, 3 and 5 joined to the 40S small ribosomal subunit. eIF2 bound to GTP and the initiator tRNA joins the ternary complex to form the 43S pre-initiation complex (43S PIC). The 43S PIC is then bound to an activated mRNA to form the 48S PIC. The 48S PIC performs scanning until the Kozak sequence is found. The 48S PIC is then joined by the 60S large ribosomal subunit, signaling the end of initiation and the start of elongation. (b) Demonstrates how translational efficiency is regulated negatively through phosphorylation of eIF2. eIF2 is bound to GTP, which is hydrolyzed during initiation. The eIF2α subunit of eIF2 can be phosphorylated at Serine 51 (S51) to negatively regulate its role in translation. Unphosphorylated eIF2 can initiate translation normally, whereas phosphorylated eIF2α prevents exchange of GDP for GTP, inhibiting initiation. (c) shows how members of the eIF4F complex are regulated by mTOR. Phosphorylation by various kinases of the mTOR signaling cascade enhances translational efficiency through inhibition of 4E-BP, as well as enhancing association of eIF4B with eIF4F
2.1.4 Initiation: Assembly of the Pre-initiation Complex
Translation initiation requires at least 12 different initiation factors that act to bring together the ribosome subunits on the mRNA and, as the most complex stage of translation, this process is highly regulated. First, the small ribosomal subunit must locate the initiation codon, facilitated by base-pairing with the anticodon of the tRNAi. Initiation begins with the formation of a pre-initiation complex (Fig. 1.1a). First, the 40S small ribosomal subunit associates with initiation factors eIF1, eIF1A, eIF5, eIF3 and the eIF2 ternary complex (composed of GTP-bound eIF2 and met-tRNAi) to form the 43S pre-initiation complex (PIC). The 43S PIC is then joins an activated mRNA to form the 48S PIC, mediated by many protein and RNA contacts including those formed between eIF4F, PABP and mRNA with the multisubunit initiation factor, eIF3. Once these translation factors have assembled on the mRNA, the next step is to locate the proper initiation codon (Aitken and Lorsch 2012; Jackson et al. 2010).
2.1.5 Initiation: Scanning for the Initiation Codon
Once the 48S PIC is assembled, the ribosome must locate the translation start site (typically the first AUG codon) to initiate protein synthesis. In order for the 48S PIC to search for the AUG start codon, it must traverse the 5′ UTR in a process known as ribosome scanning. 5′UTRs frequently contain RNA structures which can impede scanning and thus inhibit translation. In the event that the first AUG codon has a poor context, downstream AUG codons can be utilized to initiate translation, a process referred to as leaky scanning (Hinnebusch 2014).
Scanning through RNA structure by the 48S PIC is promoted by the eIF4A protein, which is an ATPase/helicase that can unwind secondary structure in the 5′UTR . Other helicases may also facilitate scanning. This process requires energy in the form of hydrolysis of adenosine triphosphate (ATP). eIF4B binds single stranded RNA (ssRNA) and also helps in unwinding. eIF4G is involved by facilitating the association of eIF4A (Hinnebusch 2014).
The factors eIF1, eIF1A, and eIF3 aid in scanning by stabilizing the open conformation of the mRNA entry channel of the small ribosome subunit, and also in start codon recognition. The 48S PIC slides along the mRNA, sampling the RNA until the first AUG codon in the proper sequence context is located. Once the AUG start codon successfully base pairs with its anticodon complement on the Met-tRNAi, eIF1 is released and the PIC adopts a more closed conformation. At this point, the Met-tRNAi is positioned in the P-site of the ribosome. The GTP bound to eIF2 is then hydrolyzed and eIF5 and GDP-bound eIF2 are released from the PIC. eIF1A is the only initiation factor from the PIC which remains bound throughout the entire process of initiation. The PIC is now more stably bound to the mRNA and tRNAi and poised for joining of the 60S subunit (Hinnebusch 2014).
2.1.6 Initiation: Formation of the 80S Ribosome
The next phase of initiation is assembly of the 80S ribosome through joining of the 60S subunit to the initiation codon associated 48S PIC. The 60S large ribosomal subunit first assembles with the GTPase protein eIF5B. Upon large and small ribosome subunit joining, eIF5B hydrolyzes its GTP. eIF5B and eIF1A are then released as the ribosome undergoes a conformational change. The resulting 80S ribosome is thereby primed to enter the elongation phase (Aitken and Lorsch 2012; Jackson et al. 2010).
2.1.7 Elongation
Once the 80S ribosome has assembled at the initiation site, protein synthesis can commence through ribosome-catalyzed peptide bond formation between the Met-tRNAi located in the P-site and the incoming amino-acylated tRNA in the A-site. The nascent polypeptide chain is extended through sequential rounds of peptide bond formation and translocation of the ribosome along the mRNA. Subsequent amino acid additions are specified through complementary base-pairing between tRNA anti-codons and the triplet codons of the mRNA. Elongation in eukaryotes is mediated by two elongation factor proteins: eEF1 and eEF2 (Dever and Green 2012). eEF1 is a multisubunit complex that delivers the amino-acylated tRNA to the ribosome (Sasikumar et al. 2012). Upon proper positioning of the tRNA in the A-site, the eEF1A subunit hydrolyzes GTP and eEF1 dissociates from the ribosome. The eEF2 factor facilitates the translocation of the ribosome and hydrolysis of GTP (Dever and Green 2012).
Typically, multiple ribosomes are sequentially assembled on and traverse the mRNA simultaneously. As the first ribosome elongates away from the initiation site, new ribosomes can initiate and follow. The resulting mRNA with multiple associated ribosomes is referred to as a poly-ribosome or polysome (Slayter et al. 1963; Warner et al. 1963). The density of ribosomes on an mRNA is proportional to the length of the open reading frame and the rates of initiation and elongation (Ingolia 2014).
2.1.8 Termination
Translating ribosomes traverse the mRNA until they encounter a stop codon (UAA, UAG, and UGA) within the A-site, which signals the termination of polypeptide chain elongation. Since there is no tRNA anticodon complementary to the stop codon, no amino acid can be added to the end of the peptide chain. Instead, a release factor (eRF) binds the stop codon and triggers the release of the complete polypeptide from the ribosome. Eukaryotes have two release factors: eRF1, which is involved in stop codon recognition and hydrolysis of the nascent protein from the P-site bound tRNA, and eRF3, a GTPase which promotes polypeptide release. Upon termination of translation, the ribosome is disassembled into its large and small subunits, assisted by additional translation factors (Dever and Green 2012; Jackson et al. 2012).
2.2 Regulation of Translation
Translation can be regulated in multiple steps to control the amount, timing and location of protein synthesis. Cis-acting sequence elements and trans-acting factors can either activate or repress translation. Translation can be regulated globally, affecting protein synthesis from all mRNAs (as is the case with the mTOR pathway), or specifically from certain mRNAs (as with sequence-specific RNA-binding proteins and microRNAs ). Groups of mRNAs can be translationally regulated in a coordinated fashion by common cis-elements and trans-factors (Abaza and Gebauer 2008; Jackson et al. 2010). One classic example of this type of post-transcriptional regulation is the 5′ terminal oligo-pyrimidine (TOP) mRNAs that encode multiple components of the translation apparatus and are coordinately regulated in response to stress (Meyuhas and Kahan 2015). Here, we will discuss control of translation and explore some of its general mechanisms (Fig. 1.2).
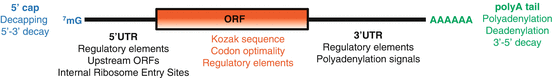
Fig. 1.2
mRNAs contain cis-acting regulatory information that controls translation efficiency and stability. Eukaryotic mRNAs have a 5′ 7-methylguanosine (7mG) cap that promotes translation and stability. The poly(A)denosine (poly(A)) tail is, recognized by poly(A) binding proteins PABP , promotes translation and stability. Removal of the cap and poly(A) tail result in subsequent mRNA degradation . Cis regulatory elements can be contained in the 5′ and 3′ untranslated regions (UTRs ) of an mRNA. These can include binding sides of RNA binding proteins and non-coding RNAs that modulate stability and translation. The open reading frame (ORF) can also contribute to regulation through the Kozak sequence and codon optimality (how commonly the codons it contains are found in the cell)
2.2.1 Regulation of Translation Initiation Through the Closed Loop
The closed loop conformation of mRNAs promotes translation initiation through the cap-to-poly(A) tail interactions mediated by eIF4F and PABP . Formation of the closed loop represents an important regulatory stage. Trans-acting factors can disrupt the closed loop to inhibit translation by displacing eIF4F or PABP from the mRNA or by disrupting their protein–protein interactions (Kawahara et al. 2008; Weidmann et al. 2014; Zekri et al. 2013). Enzymatic removal of the 5′ cap structure or poly(A) tail can disrupt closed loop formation, thereby silencing translation and leaving the mRNA vulnerable to degradation, as described below. The length of the poly(A) tail can modulate translation like a rheostat, with longer poly(A) tails promoting protein expression whereas shortening of the tail reduces it. As such, factors that stabilize or shorten the tail can control translation efficiency (Goldstrohm and Wickens 2008).
Translation efficiency can be controlled by proteins that interact with PABP . PABP-interacting proteins (PAIP1 and PAIP2) bind to PABP to either stimulate or inhibit translation, respectively (Khaleghpour et al. 2001; Roy et al. 2002). PAIP1 shares homology with eIF4G and forms a complex with initiation factors eIF4A and eIF3 to enhance translation by bridging PABP’s interaction with eIF4F and stabilizing the closed loop (Craig et al. 1998; Martineau et al. 2008). PAIP2 competes with eIF4G and PAIP1 to bind PABP, reducing its affinity for the poly(A) tail and reducing translation efficiency (Khaleghpour et al. 2001).
The 5′ cap and associated eIF4F complex are major targets for translational control mechanisms. Proteins that compete with eIF4F for binding to the 5′ cap can inhibit translation (Cho et al. 2005, 2006). A second, widely utilized control mechanism is mediated by proteins that directly bind to cap-binding eIF4F subunit, eIF4E. These eIF4E Binding Proteins (4E-BPs) competitively bind to the same region of eIF4E as the eIF4G subunit. In doing so, 4E-BPs disrupt eIF4F and the closed loop, thereby inhibiting formation of the PIC. In addition to repressing translation, several 4E-BPs have been shown to promote degradation of mRNAs (Andrei et al. 2005; Blewett and Goldstrohm 2012a, b; Igreja and Izaurralde 2011; Rendl et al. 2012).
Multiple signaling pathways intersect on 4E-BPs, providing a nexus for controlling translation. Unphosphorylated 4E-BPs have a high affinity for eIF4E whereas phosphorylation of 4E-BPs prevents their interaction with eIF4E (Fig. 1.1c). The Target of Rapamycin (TOR ) pathway is a major regulator of translation that responds to the availability of nutrients and amino acids. TOR is an important regulator of cell growth and proliferation and is inhibited in response to stress conditions and starvation. TOR also integrates signals from hormones such as Insulin and Brain-Derived Neurotrophic Factor. In turn, TOR pathway regulates translation of peptide hormones such as Leptin. TOR promotes translation in several ways, including phosphorylation of 4E-BPs and activation of S6 kinase, which phosphorylates the small ribosomal subunit 6 and eIF4B, among other targets, to promote translation (Dennis et al. 2012; Ma and Blenis 2009; Tavares et al. 2015). This cascade of TOR signaling controls translation initiation on a broad level.
2.2.2 Regulation of Initiation Through Initiation Factor eIF2
Translation initiation depends on delivery of Met-tRNAi to the 40S subunit by GTP-bound eIF2 and subsequent PIC formation. Thus, eIF2 represents an important regulatory target. eIF2 is inhibited by phosphorylation at Serine 51 (S51) on the α subunit by various kinases in response to diverse signals (Baird and Wek 2012) (Fig. 1.1b). Kinases that phosphorylate eIF2 include: (1) PKR-like endoplasmic reticulum kinase (PERK), which is activated by the unfolded protein response; (2) General Control Nonderepressible 2 (GCN2), which is activated by diverse stressors, such glucose and amino acid starvation; (3) Protein Kinase R (PKR), which is activated by dsRNAs greater than 30 bp in length and plays an important role in anti-viral response; and (4) Heme-regulated inhibitor kinase (HRI) in erythroid cells, which is activated in response to heme deficiency (Baird and Wek 2012; Lemaire et al. 2008). Phosphorylation at S51 prevents exchange of GDP for GTP, thus the phosphorylated eIF2 cannot enter new rounds of translation. As a result, translation initiation is inhibited globally (Baird and Wek 2012).
2.2.3 RNA Binding Proteins Regulate Translation
The untranslated regions (UTRs ) of many mRNAs can contain important regulatory information that controls translation (Mignone et al. 2002). RNA binding proteins (RBPs) often recognize these regions to regulate the translational efficiency and stability of target mRNAs. RBPs serve many important biological roles where gene expression needs to be quantitatively, temporally and/or spatially controlled, such as in response to hormone mediated signaling. RBPs can bind to specific RNA structures, (e.g. stem-loop structures), or they can bind to specific single-stranded sequence motifs. Upon binding to a transcript, RBPs can use diverse mechanisms to modulate translation by either repressing or activating protein synthesis (Abaza and Gebauer 2008). Here, we will explore some specific mechanisms of RBP translational repressors and activators.
RBP repressors can inhibit initiation by binding to the 5′ UTR of a target mRNA and blocking assembly of the PIC. A classic example of this mechanism is the Iron Response Protein (IRP), which, in response to low intracellular iron, binds to specific RNA stem-loop structure in the 5′UTR of ferritin mRNA, the Iron Response Element, to impede 43S joining and thus represses translation of ferritin (Muckenthaler et al. 1998).
RBP repressors can also bind to the 3′UTR of transcripts to control translation. One mechanism is to prevent assembly of the 80S ribosome. For instance, the RBPs hnRNP-K and hnRNP-E1 repress lipoxygenase mRNA by preventing 60S subunit joining to the 48S PIC (Ostareck et al. 2001). Other 3′UTR-bound RBPs can recruit 4E-BPs to a specific message to disrupt the closed loop and repress translation; examples of such interactions are numerous and include Bruno and Cup (Nakamura et al. 2004), Smaug and Cup (Nelson et al. 2004), and Puf5 and Eap1 (Blewett and Goldstrohm 2012a). This mechanism is illustrated by the RBP called Cytoplasmic Polyadenylation Element Binding Protein (CPEB ), which binds to U-rich sequences known as Cytoplasmic Polyadenylation Elements (CPEs) in the 3′UTR of certain mRNAs. One mechanism of CPEB repression is recruitment of the 4E-BP Maskin to inhibit translation of specific mRNAs, such as Cyclin B, during oogenesis (Groisman et al. 2000; Stebbins-Boaz et al. 1999).
Translation initiation can be inhibited by RBP-mediated recruitment of an eIF4E Homologous Protein (4EHP) that competes with eIF4E for binding to the mRNAs 5′ cap (Rom et al. 1998). However, unlike eIF4E, 4EHP does not interact with eIF4G and thus prevents translation initiation. The 3′UTR binding protein Bicoid recruits 4EHP to repress translation of specific mRNAs during Drosophila embryonic development (Cho et al. 2005).
RBPs can repress translation of specific mRNAs by causing shortening of the mRNAs poly(A) tail—a process referred to as deadenylation —thereby reducing or eliminating the occupancy of PABP to diminish translation initiation. One of the first examples of deadenylation mediated silencing was the finding that cytokine and growth factor mRNAs contained Adenine-Uridine Rich Elements (AREs) in their 3′UTRs which accelerated deadenylation and mRNA degradation , limiting protein expression (Wilson and Treisman 1988). These AREs can be bound by several RBPs, including the repressive Tristetraprolin (TTP ) protein which binds and recruits a multisubunit complex of poly(A) degrading enzymes that shorten the poly(A) tail of TTP bound mRNAs (Lykke-Andersen and Wagner 2005). Likewise, members of the Pumilio and Fem3 Binding (PUF) family of sequence-specific RBPs bind to 3′UTRs and recruit specialized poly(A) degrading enzymes that remove the poly(A) tail to repress protein expression (Goldstrohm et al. 2006; Van Etten et al. 2012). CPEB , as mentioned earlier, also promotes deadenylation of the mRNAs to which it binds by recruiting the poly(A) specific ribonuclease (PARN ), contributing to translational repression (Kim and Richter 2006).
3′UTR -bound RBPs can also repress translation by promoting removal of the message’s 5′ cap structure. The TTP protein interacts with and recruits decapping enzymes to specific transcripts that contain ARE sequences in their 3′UTRs (Fenger-Gron et al. 2005; Lykke-Andersen and Wagner 2005). One PUF protein can promote decapping of mRNAs by using a 4E-BP to disrupt eIF4F and to recruit decapping factors to the message, resulting in translational repression and mRNA degradation (Blewett and Goldstrohm 2012a, b). Through these mechanisms, RBP mediated translational repression and mRNA degradation are directly interrelated, a subject that we shall revisit in subsequent discussion of mRNA decay pathways in post-transcriptional control.
2.2.4 RBP Activators
Translation can also be activated by cis- and trans-acting factors, which can boost the amount of protein produced by an mRNA. They can also reanimate mRNAs that have been stored in a quiescent status, a common event in developmental contexts (Gray and Wickens 1998; Ivshina et al. 2014). Just as the poly(A) tail is a target for repressive mechanisms, it can also be employed to activate mRNAs. Polyadenylation (that is, lengthening of the poly(A) tail) and the resulting increased recruitment of PABP can promote translational activation . Thus, dormant, deadenylated mRNAs can be activated by polyadenylation in the cytoplasm via recruitment of poly(A) polymerase enzymes, such as GLD2 (Ivshina et al. 2014).
Perhaps the best characterized example of polyadenylation mediated activation is the sequence-specific RBP CPEB (Charlesworth et al. 2013). As described earlier, CPEB represses mRNAs via deadenylation and Maskin -mediated inhibition of eIF4F. CPEB acts as a bifunctional regulator during oogenesis, switching from repression to activation in response to signal the steroid hormone progesterone (Groisman et al. 2002; Ivshina et al. 2014; Sarkissian et al. 2004). Aurora A kinase phosphorylates CPEB, thereby switching it to an activation mechanism wherein CPEB interacts with and recruits GLD2 poly(A) polymerase. CPEB-Gld2 mediated polyadenylation requires that the mRNA contain both a CPE and the polyadenylation element (AAUAAA) at the 3′ end of the mRNA. The polyadenylation element is recognized by a cytoplasmic version of the Cleavage and Polyadenylation Specificity Factor (CPSF). The CPEB-GLD2 complex extends the poly(A) tails of the target mRNAs and increases the occupancy of poly(A) binding proteins. Thus, CPEB activation includes derepression and polyadenylation resulting in increased efficiency of translation (Ivshina et al. 2014).
2.2.5 Regulation of Translation Elongation
The process of elongation is iterated with an average rate of 6 amino acid additions per second (Ingolia et al. 2011). Global analyses suggest that protein synthesis rates vary over a wide range (Schwanhausser et al. 2011). Various factors impinge on the elongating ribosome to influence its speed and the location and quality of protein expression. For instance, elongation rate can be influenced by synonymous codon usage and the availability of the necessary amino-acylated tRNAs (Pechmann and Frydman 2013; Presnyak et al. 2015; Quax et al. 2015; Tarrant and von der Haar 2014). For membrane bound and secreted proteins such as hormones, a signal peptide in the nascent polypeptide is recognized by the Signal Recognition Particle to direct the translating mRNA to the proper intracellular location. Chaperones (for example, heat shock proteins) associate with and fold the nascent peptide cotranslationally (Jha and Komar 2011). In several examples, signal transduction pathways have been shown to target elongation factors to influence the rate of protein synthesis (Dever and Green 2012; Sasikumar et al. 2012).
2.2.6 Alternative Mechanisms of Initiation: Cap-Independent Translation Initiation
Translation generally requires the 5′ cap; however, in specialized instances, translation can initiate in a cap-independent manner, mediated by internal ribosome entry sites (IRES ). IRES are highly structured elements present in the 5′UTR of specific mRNAs which can allow translational initiation on that mRNA without the requirement of the 5′ cap and certain initiation factors through complex interactions with the ribosome, circumventing the process of scanning. IRES were first discovered in viral mRNAs, but examples of cellular IRES-containing mRNAs have emerged. These alternative initiation mechanisms permit translation of specific proteins when cap-dependent translation is turned off by the cell in response to viral infection, or other cellular stresses (Hellen and Sarnow 2001).
One well known example of a viral IRES -containing mRNA is that of the Hepatitis C Virus, which contains both 5′ and 3′ IRES elements (Fraser and Doudna 2007). An extreme example of a viral 5′ IRES is that the Cricket Paralysis Virus, which is able to bypass the requirement for all translation initiation factors (including eIF2-tRNAi) by mimicking the initiator tRNA in the P site of the ribosome (Fernandez et al. 2014; Hellen and Sarnow 2001). A well-studied example of a cellular IRES-containing mRNA which will be discussed in Chap. 8 is Vascular Endothelial Growth Factor A (VEGF -A), a mitogen and important stimulator of angiogenesis (Akiri et al. 1998; Huez et al. 1998; Miller et al. 1998).
3 Regulation of Gene Expression by mRNA Degradation
All mRNAs undergo decay as a part of normal gene expression. Decay of individual mRNAs can be highly regulated to control proper levels of protein expression and to spatially and temporally restrict protein production. Further, intrinsic or extrinsic signals can alter the decay rates of specific transcripts, including endocrine signals that alter gene expression by effecting mRNA decay . In this section, we provide an overview of mRNA decay pathways in mammals. We then discuss mechanisms of post-transcriptional regulation through RNA decay , including important gaps in current knowledge.
RNA decay pathways are initiated from either the 3′ or 5′ ends of the transcript, or by endonucleolytic cleavage. Multiple pathways can overlap in order to efficiently degrade an mRNA. First, we consider a major pathway of transcript decay that proceeds through the processes of deadenylation , decapping and exonucleolytic decay (Fig. 1.3).
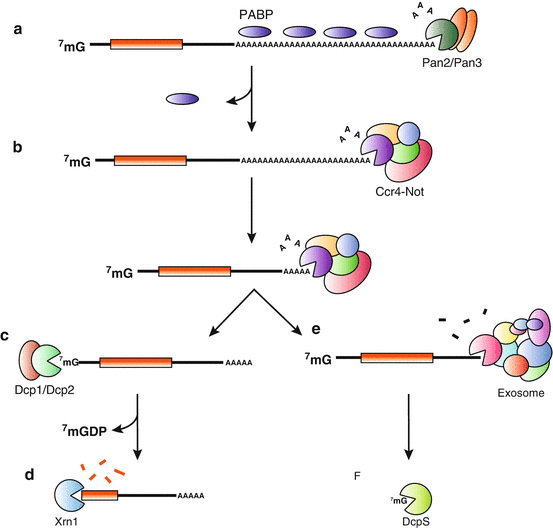
Fig. 1.3
Eukaryotic mRNA decay pathways. Decay of mRNA in eukaryotes begins with deadenylation by Pan2-Pan3 (a), which shortens the poly(A) tail and may dissociate PABP . The remainder of the poly(A) tail is degraded by the Ccr4-Not complex (b). Both deadenylase complexes release AMP molecules as the product of the deadenylation reaction. After deadenylation, mRNAs go through one of two pathways to decay the remainder of the mRNA. The 5′ to 3′ decay pathway starts with decapping by Dcp1/Dcp2 to remove the 7-methylguanosine cap (c) and is concluded by degradation of the remainder of the message by Xrn1 (d). The 3′ to 5′ decay pathway comprises of degradation of the RNA down to the cap by the Exosome (e) and subsequent scavenging of the cap by DcpS (f)
3.1 Deadenylases Remove the Poly(A) Tail
Decay of cytoplasmic mRNAs typically initiates by the progressive shortening of the 3′ poly(A) tail, a process referred to as deadenylation (Fig. 1.3). Deadenylation is frequently the rate-limiting step of mRNA decay and is an important node for control of translation and mRNA decay. Multiple enzymes catalyze deadenylation (Goldstrohm and Wickens 2008). These deadenylases are magnesium-dependent exoribonucleases that degrade mRNAs from the 3′ to 5′ end of the poly(A) tail, releasing 5′ AMP as a product. General mRNA deadenylation occurs in two phases. Once the poly(A) tail is shorted to a critical threshold, the mRNA is rapidly destroyed.
The first phase is comprised of an initial shortening of the poly(A) tail by the Pan2-Pan3 deadenylase complex (Boeck et al. 1996; Brown et al. 1996; Yamashita et al. 2005). The Pan2 subunit contains a DEDD type nuclease domain that catalyzes deadenylation . The activity of Pan2-Pan3 is stimulated by Poly(A) Binding Protein (PABP ) (Uchida et al. 2004). This interaction occurs through a specific protein interaction motif located on Pan3 (Siddiqui et al. 2007). PABP therefore serves a dual role: during translation, PABP stimulates translation initiation, and it also participates in the first phase of mRNA decay . Pan2-Pan3 also has a WD40 protein–protein interaction domain, suggesting that other protein partners may contact and regulate Pan2 activity. In support of this idea, Pan2 is known to associate with at least one RNA binding regulatory complex (Christie et al. 2013; Huntzinger et al. 2013; Kuzuoglu-Ozturk et al. 2012). Additionally, Pan2 has a catalytically dead ubiquitin hydrolase domain, though the function of this domain remains enigmatic. The Pan3 subunit contains a pseudokinase domain and zinc finger domain, both of which have been hypothesized to contribute to RNA binding (Jonas et al. 2014; Schafer et al. 2014; Wolf et al. 2014).
The second phase of deadenylation is catalyzed by the multisubunit CCR4-NOT deadenylase complex (CNOT) (Yamashita et al. 2005). Originally discovered in yeast, the CNOT complex contains two active deadenylase subunits, orthologs of the Ccr4 and Pop/Caf1 proteins. In mammals, the Ccr4 orthologs include CNOT6 and CNOT6L proteins while the Pop2 orthologs include CNOT7 and CNOT8 proteins. The CNOT complex includes at least seven other components with various functions associated with regulation and coordination of deadenylase activity, in addition to functions beyond deadenylation (Collart 2003; Tucker et al. 2001). In contrast to Pan2-Pan3, which is stimulated by PABP , addition of PABP in in vitro experiments inhibits the activity of CNOT (Tucker et al. 2002). CNOT deadenylates the poly(A) tail down to a short oligo(A) that is incapable of binding PABP to facilitate translation initiation. Thus, PABP and CNOT have opposing stimulatory and inhibitory activities with respect to translation, placing poly(A) at the nexus of control of translation and mRNA decay .
The CNOT complex is regulated by protein interactions with multiple partners (Goldstrohm and Wickens 2008). For example, multiple members of the PUF family of sequence specific RNA binding proteins have been shown to bind the Pop2 subunit and recruit the CNOT complex to specific mRNAs (Goldstrohm et al. 2006, 2007; Weidmann et al. 2014). As a consequence, the poly(A) tails of the mRNAs are removed more quickly, the resulting deadenylated mRNA is translationally repressed, and its stability is decreased. A variety of RBPs have now been shown to recruit the CNOT complex to achieve repression, including the ARE binding protein TTP (Lykke-Andersen and Wagner 2005; Sandler et al. 2011) and the RBP Roquin (Leppek et al. 2013), both of which cause repression and degradation of cytokine mRNAs. RBPs can recruit the complex via interactions with specific subunits. The CNOT1 subunit is important for deadenylation , though it has no catalytic function itself. Instead, CNOT1 serves as a molecular scaffold and a target for RBP mediated recruitment to specific substrate mRNAs. For instance, TTP binds directly to CNOT1. Other CNOT complex subunits play additional functional roles, as recently reviewed by Shirai et al. (2014).
Other deadenylases have also been identified in mammalian systems. The presence of multiple deadenylases could lead to redundancy in their function, yet certain deadenylases have been shown to have specific biological functions such as control of development, anti-viral response, and regulation of metabolism (Goldstrohm and Wickens 2008). Also, some deadenylases have unique activities and features. The PARN deadenylase, expressed in many higher eukaryotes, is unique in that it binds to the 5′ cap of mRNAs, resulting in enhancement of PARN’s ability to deadenylate the mRNAs 3′end (Gao et al. 2000; Martinez et al. 2000). This property indicates that PARN likely acts on translationally inactive mRNAs. PARN has been implicated in developmental processes in plants and Xenopus laevis and is also expressed in mammals. Of particular interest in the field of endocrinology, the deadenylase Nocturnin is controlled by the circadian clock (Green and Besharse 1996), and genetic analysis indicates that Nocturnin’s main function is to regulate fat metabolism (Green et al. 2007). Still, much remains to be elucidated with regard to the functions of deadenylase family members and their roles in post-transcriptional regulation.
3.2 Decapping Enzymes Catalyze Removal of the 5′ Cap
In addition to its role in translation initiation, the 5′ 7-methylguanosine cap protects the 5′ end of the mRNA from exonucleases. These features put the 5′ cap at the nexus of regulation of translation and mRNA decay . The 5′ cap can be removed by hydrolysis of the 5′ to 5′ triphosphate linkage that covalently joins the 7-methyl guanosine to the first nucleotide of the mRNA. This process is referred to as decapping and is catalyzed by specialized decapping enzymes. Decapping typically follows deadenylation , coordinated by protein interactions between the deadenylase complex and the decapping machinery. However, deadenylation independent decapping has also been observed for some mRNAs (Badis et al. 2004; Fromont-Racine et al. 1993).
Multiple decapping enzymes have been identified (Li and Kiledjian 2010; Song et al. 2013). One of the best-characterized decapping enzymes is the highly conserved Dcp2 enzyme, which releases 7mGDP from the mRNA 5′ end through activity of its Nudix domain (Wang and Kiledjian 2002). Dcp2 also possesses a Box A domain that functions in RNA binding.
Decapping enzymes are highly regulated by protein partners (Jonas and Izaurralde 2013). Dcp2 activity is stimulated by its protein partner Dcp1, which forms a stable complex with Dcp2 through the Dcp2 Box A domain (Piccirillo et al. 2003; She et al. 2006). Dcp2 is additionally activated by Dhh1, an RNA helicase, and Pat1, which recruits the Lsm complex. (Ling et al. 2011). The Lsm protein complex (Lsm 1-7) associates with the 3′ deadenylated end of mRNA and increases Dcp2 decapping efficiency (Tharun and Parker 2001). An additional decapping factor, Ge-1, is found in this complex in higher eukaryotes, further activating decapping activity (She et al. 2008; Yu et al. 2005).
RNA binding proteins greatly influence decapping rates of specific mRNAs, as explored below and in several recent reviews (Arribas-Layton et al. 2013; Li and Kiledjian 2010). For instance, the ARE -binding protein TTP recruits decapping factors to accelerate decapping of specific transcripts (Gao et al. 2001; Lykke-Andersen and Wagner 2005). Decapping of mRNAs is frequently the fate-determining step that targets mRNAs for destruction; however, recent evidence indicates that some transcripts may be recapped, suggesting that decapping and recapping could serve as a regulatory mechanism to repress and then activate mRNAs (Mukherjee et al. 2012).
3.3 Exoribonucleolytic Decay Can Initiate from the 5′ End
Following decapping , the exposed 5′ end of the mRNA can be attacked by exoribonucleases, degrading the mRNA in a 5′ to 3′ direction while releasing nucleotide monophosphate products. Several 5′ exoribonucleases have been identified in eukaryotes including XRN1 and XRN2 (Nagarajan et al. 2013). Both enzymes act on multiple types of substrate RNAs. Here we consider several important roles.
The exoribonuclease XRN1 is primarily responsible for 5′ mRNA decay in the cytoplasm. XRN1 has specificity for RNAs with a 5′ monophosphate, coinciding with the product of DCP2-mediated decapping . This enzyme is fast and highly processive; decay intermediates are rarely detected (Nagarajan et al. 2013).
XRN2, a 5′ to 3′ exoribonuclease with homology to XRN1, is primarily located in the nucleus and is conserved across a range of species (Miki and Grosshans 2013). Functions of XRN2 include maturation of rRNA and snoRNAs, as well as transcription termination (Boisvert et al. 2007; Luo et al. 2006; Wang and Pestov 2011). XRN2 is also involved in RNA quality control pathways, decaying aberrant RNAs in the nucleus, as well as unspliced pre-mRNAs to control mRNA levels (Das et al. 2003).
3.4 Exoribonucleolytic Decay from the 3′ End
Messenger RNAs can also be degraded from the 3′ end by the exosome, a large multisubunit complex (10–12 subunits) which acts in a 3′ to 5′ direction. Two variations of the exosome complex have been characterized: one nuclear exosome and one cytoplasmic exosome. The exosome acts to process and/or degrade multiple types of RNA including mRNAs (Liu et al. 2006). The cytoplasmic exosome acts on mRNAs that have been deadenylated, degrading them from the 3′ end to produce nucleotide monophosphates. The Dis3 subunit is responsible for this activity (Reis et al. 2013). Once the mRNA is degraded to a capped m7GpppN (where N is fewer than 10 nucleotides) product, a specialized scavenger decapping enzyme, DcpS, hydrolyzes the cap structure to produce 7-methylguanosine monophosphate and nucleotide diphosphate (Chen et al. 2005; Liu and Kiledjian 2005). DcpS action prevents the potentially toxic effects of accumulated capped mRNA fragments, which could competitively inhibit eIF4F function. Interestingly, defects in this final mRNA decay step cause a form of intellectual disability and neuromuscular disease (Ng et al. 2015).
As we have learned, mRNAs can be degraded from the 5′ end, the 3′ end, or both. The 5′ and 3′ decay pathways appear to compete to degrade certain mRNAs, whereas other mRNAs appear to be mainly degraded by one or the other pathway. The determinants and factors that affect these destructive choices remain incompletely understood.
3.5 Endonucleases Cut mRNAs to Initiate Decay
As an alternative pathway to exoribonucleolytic decay, select messages undergo endonucleolytic cleavage (Fig. 1.4). The resulting 5′ and 3′ fragments are subsequently degraded by exoribonucleases including XRN1 and the exosome. In fact, the Dis3 subunit of the exosome is also an endonuclease, in addition to its exoribonuclease activity (Arraiano et al. 2010). Other examples of endonucleases highlight the diversity within this class of enzymes. One of the first characterized mammalian endoribonucleases was the PMR1 enzyme, which was found to be an estrogen induced factor that initiates mRNA decay (Pastori et al. 1991a, b). PMR1 associates with polysomes to efficiently degrade specific mRNAs. Recent evidence indicates that PMR1 has an important role in reducing parathyroid hormone mRNA levels, discussed further in the chapter by Naveh-Many (Nechama et al. 2009). The Regnase-1 endonuclease regulates immune function by degrading pro-inflammatory cytokine mRNAs (Matsushita et al. 2009) (Fig. 1.4). Regnase-1 is rapidly degraded upon stimulation of immune responses, and in mouse models, Regnase-1 is implicated in autoimmune disorders (Liang et al. 2010).
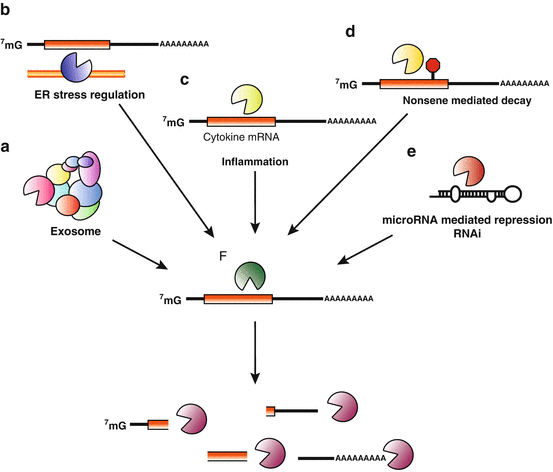
Fig. 1.4
Endonucleolytic decay pathways. Representative examples of eukaryotic endonucleolytic decay pathways are illustrated here. Components of the exosome, such as Dis3, have endonuclease activity (a). Under ER stress conditions, Ire-1 decays ER-associated mRNAs to reduce translation (b). Pro-inflammatory mRNAs are degraded by Regnase-1 to negatively regulate cytokine expression (c). In nonsense mediated decay, mRNAs with premature stop codons are cleaved by SMG6 before being degraded by exonucleases (d). In microRNA -mediated decay pathways and RNAi pathways, precursor miRNAs are processed by endonucleases such as Dicer and Drosha to make mature miRNAs (e). Endonucleolytic decay products typically serve as substrates for further decay by exonucleases (f)
Endonucleolytic mRNA decay is also an important for stress response. The IRE1 protein is a transmembrane endoribonuclease located on the endoplasmic reticulum. During stress conditions, IRE1 decays mRNAs through the regulated IRE1 dependent decay (RIDD) pathway (Hollien et al. 2009; Maurel et al. 2014) (Fig. 1.4). IRE1 substrates are specifically associated with the ER and decay faster during stress conditions to reduce translation (Gaddam et al. 2013). Additionally, these substrate mRNAs are highly enriched for transcripts involved in secretory pathways, including the hormone insulin and multiple cell surface receptors (Han et al. 2009a).
Nonsense mediated decay (NMD) of mRNAs is a vital pathway for clearing mRNA transcripts with premature termination codons. These aberrant transcripts are targeted by NMD machinery for degradation, as reviewed by Popp and Maquat (2013). One NMD component, SMG6, is an endonuclease that cleaves the mRNA, resulting in subsequent exoribonucleolytic decay of the fragments (Franks et al. 2010; Schmid and Jensen 2008) (Fig. 1.4). Endonucleases function in a variety of other RNA decay and processing pathways in the cell, including the Argonaute, Dicer , and Drosha endonucleases that participate in RNA interference pathways, addressed in a later section of this chapter.
3.6 Regulation of mRNA Decay
Cis-acting RNA sequences, either linear motifs or secondary structures, can control decay of the transcript. These elements are recognized by sequence specific RNA binding regulatory factors. In turn, the regulators recruit the mRNA decay machinery to facilitate removal of the poly(A) tail, the 5′ cap, or to promote exonucleolytic or endonucleolytic decay. There are many hundreds of RNA binding proteins encoded in mammalian cells (Gerstberger et al. 2014). Moreover, mRNA decay can be controlled by small non-coding RNAs that form complexes with regulatory proteins (Jonas and Izaurralde 2015), as described in subsequent sections. Here, we a provide a few examples or regulators that control mRNA stability and refer readers to recent in-depth reviews (Garneau et al. 2007; Goldstrohm and Wickens 2008; Li and Kiledjian 2010; Schoenberg and Maquat 2012).
Two well-characterized instability elements are the Adenine and Uridine Rich Elements (AREs) and Guanine and Uridine Rich Elements (GREs), which are frequently found in 3′ UTR of mRNAs such as cytokine mRNAs (Bakheet et al. 2006; Vlasova-St Louis and Bohjanen 2011). Multiple RNA binding proteins recognize these sequences to control mRNA stability . Certain ARE binding protein s can recruit mRNA decay machinery to increase decay. For instance, TTP recruits the CNOT deadenylase complex, the DCP2 decapping complex, and exonucleases to mRNAs to promote their destruction (Chen et al. 2001; Fenger-Gron et al. 2005; Lykke-Andersen and Wagner 2005; Sandler et al. 2011).
AREs can act as a bifunctional switch, causing mRNA decay in one state while stabilizing mRNAs in other conditions. This is achieved by at least two mechanisms. First, stabilizing ARE binding protein s , such as HuR , can compete with destabilizing factors such as TTP . Second, post-translational modifications can alter the activity of the ARE binding proteins (Garneau et al. 2007). Phosphorylation of TTP can alter its RNA binding, protein interactions, and protein stability (Brooks and Blackshear 2013). ARE mediated regulation will be further discussed in Chaps. 3, 5, 8, 9, 11, and 13.
GRE elements are bound by CUG-Binding protein, also referred to as CELF1 (Vlasova-St Louis and Bohjanen 2011) (see Chap. 3 of this volume). CELF1 plays multiple roles in mRNA processing, translation and stability. This evolutionarily conserved repressor represses protein expression by causing deadenylation and mRNA decay . The mechanism is incompletely understood, but evidence indicates that CELF1 interacts with the deadenylase PARN to promote deadenylation (Moraes et al. 2006).
3.7 Nonsense-Mediated, Non-stop, and No-Go mRNA Decay Pathways
Decay of mRNA is a vital point of normal gene regulation , but also plays an important role in quality control of gene expression (Ghosh and Jacobson 2010). Aberrant, defective mRNAs are degraded to ensure fidelity. These mRNAs can arise due to mutation, misprocessing, or breakdown in the complex processes necessary to decode them (i.e. translation). Nonsense mediated decay, discussed above, is responsible for decaying transcripts with premature termination codons, protecting cells from the potentially deleterious effects of producing truncated proteins with abnormal function (Popp and Maquat 2014). NMD destroys these aberrant transcripts via deadenylation dependent decay and endonucleolytic decay (Schoenberg and Maquat 2012). Messenger RNAs that lack a stop codon, as the result of mutation or misprocessing, are targeted by Non-stop decay pathway, wherein the exosome destroys the transcript. An additional quality control pathway, so-called No-Go Decay, clears faulty mRNAs stalled ribosomes stuck in the act of translation. This decay pathway releases the ribosome and then degrades the abnormal transcript via endonucleolytic cleavage. For more information on quality control mechanisms, we refer readers to recent reviews (Harigaya et al. 2010; Popp and Maquat 2013).
4 Post-transcriptional Regulation by Non-coding RNA s
The protein-coding sequence in the human genome only accounts for 1 % of the entire genome meaning that the majority of the genome is noncoding DNA (Mattick 2004). Much of this noncoding DNA is actually transcribed into noncoding RNAs (Bertone et al. 2004; Cheng et al. 2005; Kampa et al. 2004). This finding suggests that higher eukaryotes have evolved new and complex regulatory mechanisms, both structural and functional, that involve not just proteins but also noncoding RNAs. Many of these noncoding transcripts are synthesized similar to mRNAs including being capped at the 5′ end, often spliced, and polyadenylated at the 3′ end. Further processing may take place once these RNAs are in processing complexes to generate functional noncoding RNAs. The regulatory potential of RNAs comes from the ability for it to interact with other nucleic acids and proteins allowing for intricate formation of regulatory RNA-protein (RNP) complexes. Such regulatory RNAs include small noncoding RNAs such as microRNAs (miRNAs ) and short interfering RNAs (siRNAs) (Carthew and Sontheimer 2009). Long noncoding RNAs and circular RNAs are additional forms of noncoding RNA that have more recently been shown to contribute to gene regulation (Geisler and Coller 2013). These RNAs can serve many critical functional roles in the cell including control of transcription, post-transcriptional regulation of mRNA decay and translational control .
4.1 Small Non-coding RNA s
4.1.1 MicroRNAs
MicroRNAs are 22–25 nt small noncoding RNAs that participate in RNA-base pairing interactions with specific mRNAs in order to repress expression of target mRNAs (Bartel 2004). They do so by inhibiting translation and activating mRNA degradation (Jonas and Izaurralde 2015). Post-transcriptional gene regulation by microRNAs is widespread (Bartel 2004, 2009). Currently, over 1500 microRNAs have been identified in the human genome, each of which can regulate expression of multiple genes (Freedman and Tanriverdi 2013; Lewis et al. 2005). Some of these are conserved across mammals and even to lower eukaryotes, while others are endogenous to the human genome only. Many are expressed in a tissue specific manner, (Londin et al. 2015) while others are expressed in stage-specific manners during development (Krichevsky et al. 2003; Pasquinelli et al. 2000). Because of this, microRNAs are critical regulators of many tissue specific functions. For example: one pancreatic specific microRNA , miR-375, regulates several mRNA targets that are critical for the secretion of insulin from the islets of Langerhans (Poy et al. 2004). If miR-375 is lost, insulin secretion is upregulated and when miR-375 is abundant, insulin secretion is downregulated. Another microRNA, miR-143, was identified through a microarray analysis to play an important regulatory role in adipocyte differentiation (Esau et al. 2004). These are just a few examples of the numerous biological roles of microRNAs. This book will emphasize the regulatory roles of microRNAs in endocrine function, as discussed in Chaps. 2, 5, 6, 9, 12, 13 and 14.
Biogenesis of MicroRNAs
MicroRNAs are processed from precursor transcripts through several steps before they become functional. First, microRNAs are transcribed by RNA Polymerase II; these transcripts are usually capped and poly(A)denylated. Typically, these primary transcripts can encode for one or more microRNAs and can even code for clusters of microRNAs (Lagos-Quintana et al. 2001; Lau et al. 2001). Alternatively, one transcript can encode for both a microRNA and a protein simultaneously. In these cases, the microRNA sequence is many times encoded in the intronic sequence, so-called mirtrons (Berezikov et al. 2007; Ha and Kim 2014; Okamura et al. 2007).
The initial primary microRNA (pri-miRNA) has extra sequence extending past the microRNA itself (22–24 core nucleotides) both on the 5′- and 3′-ends of the transcript. Next, the pri-miRNA is folded into a stem-loop structure and is excised from the primary transcript while still in the cell’s nucleus. A complex including an RNase III endonuclease, Drosha , carries out this cleavage step. Drosha is accompanied in this complex by DGCR8, a protein that serves to recognize the pri-miRNA. The Drosha-DGCR8 complex cleaves the pri-miRNA such that a 60–70 nucleotide stem-loop intermediate with a mature 5′-phosphate and a 3′-nucleotide overhang remains. This product of Drosha cleavage is referred to as a pre-miRNA and can be exported to the cytoplasm for further processing and maturation (Lee et al. 2003; Zeng and Cullen 2003).
In the cytoplasm, the pre-miRNA becomes a substrate for Dicer , an RNase III endonuclease which catalyzes a second cleavage event. Dicer specifically recognizes the 3′-nucleotide overhang that was previously generated during Drosha processing. This endonuclease event is responsible for removal of the terminal loop and consequently the release of a small double stranded RNA duplex. This Dicer cleavage event additionally dictates the length of the duplex RNA for the mature miRNA of 22–25 nucleotides. Once the mature miRNA is formed, it can enter into a functional regulatory complex referred to as the RNA-induced silencing complex (RISC ) (Ha and Kim 2014; MacRae et al. 2007).
Regulation of MicroRNA Biogenesis
MicroRNA biogenesis can be regulated at each processing step, thereby altering the regulatory response controlled by the resulting microRNA (Ha and Kim 2014). Drosha processing activity can be modulated by DGCR8 autoregulation in the first step of microRNA processing (Han et al. 2009b). The DGCR8 mRNA contains conserved stem loop structures that closely resemble the structure formed by the pri-miRNA and therefore the Drosha/DGCR8 complex is able to cleave the DGCR8 mRNA, controlling the expression of DGCR8 at the post-transcriptional level. Furthermore, the DGCR8 component of the complex forms protein–protein interactions with Drosha in order to further stabilize Drosha creating a feedback mechanism to control the levels of this microprocessor complex (Han et al. 2009a, b). Importantly, this mechanism is highly conserved from humans to zebrafish to drosophila.
Another example of the regulation of microRNA biogenesis occurs at the stage of Dicer -catalyzed microRNA processing (Ha and Kim 2014). The best example is the let-7 microRNA and one of its canonical targets, Lin28. Lin28 is typically translationally repressed by the let-7 microRNA; however, when Lin28 protein is present, it binds to the terminal loop of the let-7 pre-miRNA leading. This interaction impedes the binding and cleavage by Dicer and thus the pre-let-7 miRNA maturation is inhibited (Carthew and Sontheimer 2009; Ha and Kim 2014). Instead, Lin28 recruits Terminal Uridyl Transferases (TUTs) to cause oligouridylation, or the addition of uridylate residues, to the 3′ end of the pre-let-7 miRNA and consequently degradation of the microRNA intermediate (Heo et al. 2012; Thornton et al. 2012). These types of mechanisms allow for tight control of microRNA production and processing and by controlling the levels of mature microRNAs , the cell is able to modulate regulation by microRNAs.
4.1.2 Short Interfering RNAs
Short interfering RNAs (siRNAs) are 21–23 nucleotide RNAs processed from exogenous double-stranded RNAs (dsRNAs). SiRNAs are fully complementary to their target mRNA sequences and work primarily by Watson-Crick base-pairing (Carthew and Sontheimer 2009). Targeting of an siRNA to an mRNA results in an endonucleolytic cleavage and subsequent exonucleolytic decay of the mRNA fragments. In research, siRNAs are often administered to cell culture for programmed knockdown of a particular gene via RNA interference (RNAi) or post-transcriptional gene silencing (PTGS). In cells, Dicer can also process double-stranded RNAs into functional siRNAs.
Another class of siRNAs, endo-siRNAs, are produced from endogenous cellular dsRNA precursors (Ghildiyal et al. 2008; Yang and Kazazian 2006). Endo-siRNAs differ from microRNAs in the ways in which they are generated and processed. These RNAs are synthesized and processed in a variety of different ways but typically require Dicer for cleavage of a double stranded RNA (dsRNA) precursor and/or RNA-dependent RNA polymerases in order to be synthesized and processed. RNA-dependent RNA polymerases can use mature mRNAs as templates for synthesis, and the products are cleaved by Dicer. The resulting endo-siRNAs can act as silencers of both transcription and translation (Ghildiyal and Zamore 2009). Endo-siRNAs produced from viral RNAs can protect the cell from viral infections (Li et al. 2013; Wang et al. 2006). Historically, there is strong evidence for this type of mechanism in plants and flies; however, more recently, these anti-viral RNAi response pathways have been observed in multiple mammalian cell lines, suggesting evolutionary conservation (Claycomb 2014; Li et al. 2013).
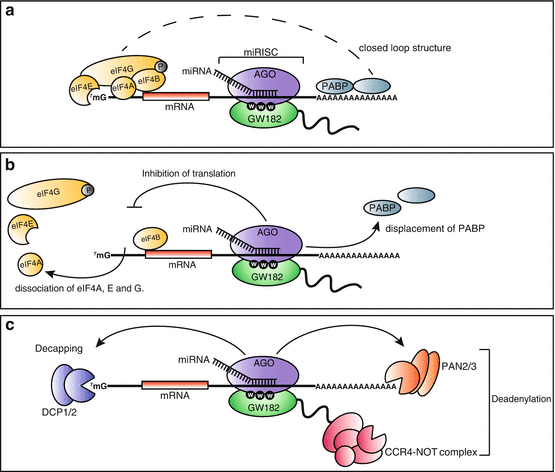
4.2 Assembly of the RNA Induced Silencing Complex (RISC )
To control gene expression, microRNAs and siRNAs must be loaded into protein complexes termed RNA induced silencing complex es (RISC ) (Jonas and Izaurralde 2015) (Fig. 1.5). A core component of RISC is the Argonaute protein family (Meister 2013). When assembled, RISC contains one of several Argonaute (Ago) proteins that bind directly to the small RNAs. Eukaryotes often possess more than one Argonaute protein that share similar architecture including a domain with homology to endonucleases (Meister 2013). There are four human Argonautes (AGO1-4), each of which can bind to small RNAs; however, only AGO2, is enzymatically capable of endonucleolytic cleavage of bound mRNA targets.
 Translational and Post-translational Control of Leptin Production by Fat Cells
Translational and Post-translational Control of Leptin Production by Fat Cells
 MicroRNA Regulation of Endocrine Functions in the Ovary
MicroRNA Regulation of Endocrine Functions in the Ovary
 Post-transcriptional and Post-translational Regulation of Steroidogenesis
Post-transcriptional and Post-translational Regulation of Steroidogenesis
 Post-transcriptional Regulation of Prostaglandin Biosynthesis
Post-transcriptional Regulation of Prostaglandin Biosynthesis
 Post-transcriptional Regulation of Insulin and Insulin Like Growth Factors
Post-transcriptional Regulation of Insulin and Insulin Like Growth Factors
 Post-transcriptional Regulation of Parathyroid Hormone Gene Expression in Health and Disease
Post-transcriptional Regulation of Parathyroid Hormone Gene Expression in Health and Disease




Related posts:
Translational and Post-translational Control of Leptin Production by Fat Cells
MicroRNA Regulation of Endocrine Functions in the Ovary
Post-transcriptional and Post-translational Regulation of Steroidogenesis
Post-transcriptional Regulation of Prostaglandin Biosynthesis
Post-transcriptional Regulation of Insulin and Insulin Like Growth Factors
Post-transcriptional Regulation of Parathyroid Hormone Gene Expression in Health and Disease
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
