Model (order determined by date of first published report)
Notes on pathophysiology
Protective interventions
Femoral vein pinch injury (mouse): injection of patient aPL, followed by a standard pinch injury to the femoral vein. The same mice may also be characterized for vessel wall activation in carotid homogenates, distant from the site of thrombosis
Patient aPL promote larger and more durable thrombi after pinch injury [24], including IgG, IgM, and IgA preparations [25]
Enhanced leukocyte adhesion in the cremaster microcirculation correlates with enhanced thrombosis in veins [26]
Carotid artery homogenates express increased tissue factor in aPL-treated mice (albeit distant from the site of thrombosis) [27]
Anti-β2GPI isolated from leprosy patients are not thrombogenic [28]
Affinity-purified antibodies specifically targeting domain I of β2GPI are thrombogenic [29]
Protective drugs/inhibitors:
Hydroxychloroquine [30]
Anti-VCAM-1 [31]
Fluvastatin [32]
Anti-C5 [33]
TIFI, a peptide that mimics domain V of β2GPI [34]
MG132, a specific NFκB inhibitor [35]
Recombinant domain I of β2GPI [36]
C5-inhibitor rEV576 [37]
Protective mutations:
ICAM-1 [31]
P-selectin [31]
E-selectin [38]
C3 [33]
C5 [33]
C5a receptor [39]
TLR4 [27]
Annexin A2 [40]
ApoER2 [41]
C6 [42]
Photochemically induced carotid thrombosis (hamster): injection of mouse monoclonal aβ2GPI, followed by Rose Bengal-mediated photochemical injury
Mouse monoclonal aPL promote arterial thrombosis in a dose-responsive fashion [43]
F(ab′)2 aPL fragments can promote thrombus formation [43]
LPS-priming, with imaging of mesenteric microcirculation (rat): Coadministration of LPS and patient aPL, followed by intravital imaging of the mesenteric microcirculation for occlusions
This is essentially the only model in the literature that does not require vessel wall manipulation to induce thrombosis [44]
Fibrin is deposited in the microcirculation when aPL and LPS are administered together or alone, neither is sufficient [44]
A functional Fc domain is required for an engineered monoclonal aβ2GPI to induce thrombosis [45]
Laser-induced vessel wall injury in cremaster arterioles (mouse): injection of patient aPL or affinity-purified aβ2GPI, followed by laser injury and intravital microscopy
aPL promote the rapid accumulation of both platelets and fibrin at the site of endothelial injury [46]
aPL/β2GPI complexes associate with platelets, more so than the endothelium, at the site of vessel wall injury [47]
Eptifibatide (an antiplatelet drug) blocks aPL-mediated thrombus formation, fibrin generation, and endothelial-cell activation [47]
F(ab’)2 aPL fragments can promote thrombus formation [47]
A1-A1 (an antagonist of aPL-ApoER2 interactions) is protective [48]
Ferric chloride application to mesenteric microcirculation (mouse): injection of patient aPL followed by exteriorization of the mesenteric microcirculation and application of ferric chloride
aPL antagonize eNOS and thereby reduce leukocyte adhesion to the endothelium [49]
eNOS−/− and ApoER2−/− mice are protected from aPL-mediated thrombosis [49]
Dorsal skinfold chambers (mouse): injection of mouse monoclonal aPL, followed by laser injury to small veins (visualized in implanted chambers)
The authors argue that aPL trigger a prothrombotic state by inducing monocyte tissue factor, resulting in thrombosis at the site of laser-induced endothelial injury [50]
The NFκB specific inhibitor DHMEQ is protective [50]
Ferric chloride-induced carotid injury (mouse): injection of patient aPL or animal aβ2GPI, followed by ferric chloride application to carotid artery (or femoral vein)
IVC flow restriction (mouse): injection of cofactor-independent human monoclonal aPL followed by narrowing of the IVC to trigger thrombosis
Cofactor-independent aPL accelerate venous thrombosis
The phenotype is not dependent on TLR4 [53]
Acceleration of thrombosis by patient aPL is dependent upon neutrophils and neutrophil extracellular traps [54]
NOX2 mutation (in the circulating compartment only) is protective
Anti-tissue factor is protective [53]
Deoxyribonuclease and neutrophil depletion are protective [54]
Most extensively characterized is a model that involves administration of patient aPL to mice, followed by application of a standardized pinch injury to the femoral vein. A microscopic thrombus then forms and resolves over approximately 10 min, aPL-treated mice consistently form larger and more durable thrombi. The reproducibility and robustness of this model are an obvious advantage, leading to an impressive number of publications [24–42]. Disadvantages include, first, that it is debatable how well discrete mechanical injury to a vein wall mimics venous thrombosis pathogenesis in patients and, second, that vessel wall characterizations in this model typically are distant from the site of injury (e.g., studying tissue factor [TF] expression in the carotid artery, while assessing thrombosis in the femoral vein). However, the strengths of the model outweigh its weaknesses, as evidenced by the multiple pathways and concepts identified for further study (in some cases confirmed in independent models), including endothelium-leukocyte interactions, toll-like receptor (TLR) pathway signaling, complement cascade, nuclear factor kappa B (NFκB)- mediated transcription, and the key role of β2GPI, among others. Manuscripts based on this model argue that activated endothelium is a critical regulator of aPL-associated thrombosis risk.
Newer models have moved from the venous circulation to the arterial, where explicit vessel wall damage is relevant. Some of these models use intravital microscopy, permitting the characterization of specific cells, such as platelets, as the earliest players in the thrombotic event [47]. Other recent studies argue for activation of circulating cells as the key contributor [51, 53], outweighing the contribution of endothelial activation. Two models deserve special note as they do not explicitly damage the endothelium but rely on either lipopolysaccharide (LPS) administration [44] or flow restriction [53] to activate the endothelium.
Cell Activation Is Key in Thrombotic Antiphospholipid Syndrome
Platelets
During the earliest days of APS research , investigators noted the frequent occurrence of thrombocytopenia in patients and animal models [55]. In patients, elevated urinary secretion of a major platelet-derived thromboxane metabolic breakdown product, 11-dehydro-thromboxane B2 (11-dehydro-TXB2), has been reported [56]. Thus in APS the link between thrombosis and platelet activation has long been suspected and was one of the first studied aspects of the disease [55]. Both epidemiological and mechanistic studies indicate that aPL induce expression of thromboxane B2 (TXB2) and fibrinogen receptor glycoprotein IIb/IIIa (GPIIb/IIIa) in platelets, resulting in platelet aggregation [57, 58], and there is evidence for platelets’ critical role in development of thrombosis in APS patients. Indeed, in an APS mouse model, B2GPI/aB2GPI complexes localize preferentially to platelets, inducing their activation at the site of arteriolar injury [47] and suggesting that at the level of microcirculation platelets may be primary targets of aPL and that their activation leads to endothelial engagement and fibrin generation. A summary of mechanisms of aPL-mediated cell activation important for thrombus development in APS is shown in Fig. 5.1.
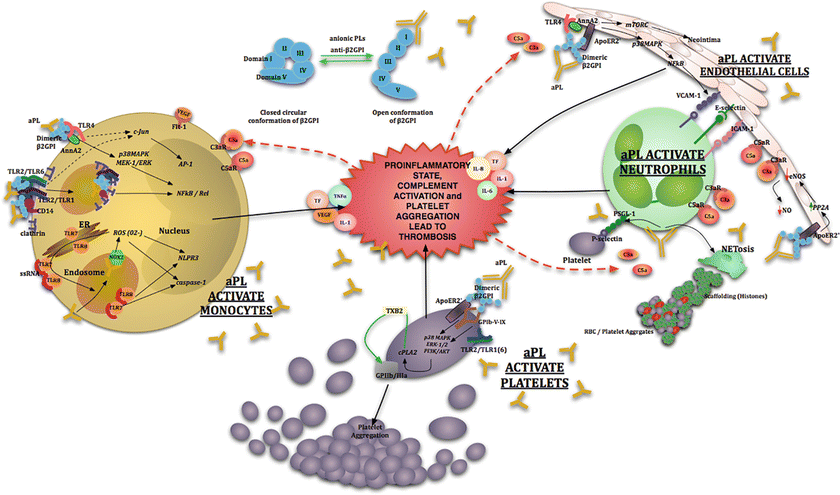
Fig. 5.1
Antiphospholipid antibody (aPL)- mediated cell activation: Pathogenic aPL activate endothelial cells (ECs) via p38 mitogen-activated protein kinase (p38 MAPK) and nuclear factor κB (NFκB) to produce several proinflammatory cytokines and adhesion molecules. Antiphospholipid antibodies can also induce activation of the mammalian target of rapamycin (mTORC) pathway in endothelial cells to induce neointima formation (i.e., endothelial hyperplasia) commonly seen in aPL nephropathy and catastrophic APS. Signaling through apolipoprotein endothelial receptor 2 (ApoER2) antagonizes endothelial nitric oxide synthase (eNOS) resulting in reduced nitric oxide production. Antiphospholipid antibodies can also activate monocytes via several signaling pathway kinases and transcription factors including p38MAPK, MEK-1/ERK, NFκB/Rel, c-Jun, AP-1, Nox2, TLR7/8 and the NLPR3 inflammasome by engagement of various target receptors to produce proinflammatory cytokines. Signal activation can occur by engagement of receptors at the cell surface or through internalization with subsequent activation of intracellular receptors. Platelet activation occurs via engagement of ApoER2, GPIbα subunit of GPIb-V-IX and/or TLR2 inducing p38 MAPK, ERK-1/2, and PI3K/AKT to induce thromboxane B2 production, GPIIb/IIIa upregulation, and platelet aggregation. Infection/inflammation plays a central role in upregulating autoantibody production, causing endothelial injury and activating monocytes and neutrophils via complement-complement receptor interactions. Antiphospholipid antibodies can induce the formation of neutrophil extracellular traps (NETs) that propagate inflammation and contribute to thrombosis
Endothelium
Given its constant interaction with whole blood, the endothelium has properties that potently counter coagulation/thrombosis [9]. The endothelium is the gateway by which inflammatory cells leave blood to enter tissue, a tightly regulated process that involves rolling, firm adhesion, and extravasation. These critical events are dependent upon selectin-mediated interactions that facilitate rolling and stronger integrin-mediated interactions that promote firm adhesion [59]. In animal models of aPL-accelerated thrombosis and in APS patients, there are signs of smoldering endothelial activation. For example, TF activity is increased in carotid homogenates from aPL-treated mice [27], a finding that correlates with increased leukocyte-endothelium interaction [26] and is supported by the facts that antagonizing E-selectin and P-selectin (key selectins expressed on endothelium) protects against thrombosis in mice and that strategies blocking the endothelial integrin ligands VCAM-1 and ICAM-1 do the same [31, 38]. Mechanistically, another study suggests that downregulation of endothelial nitric oxide synthase (eNOS) by aPL may also be an important factor increasing leukocyte-endothelium interplay [49].
In patients with APS, vascular endothelial growth factor (VEGF) and soluble TF circulate at increased levels, albeit without definitive evidence that these factors come from endothelium [60]. Kidney biopsies from patients with APS nephropathy suggest activation of the mammalian target of rapamycin (mTOR) pathway [61], which could enhance endothelial cell proliferation and certain types of APS-associated nonrenal vasculopathy, if not thrombosis. Multiple studies demonstrate endothelium-derived microparticles in the circulation of patients with APS, as a possible surrogate for endothelial activation [62, 63]. Robust in vitro evidence indicates that aPL can activate endothelial cells to express TF and adhesion molecules [64, 65]. Mechanistically, NFκB, p38 mitogen-activated protein kinase (MAPK), and Krüppel-like factors (KLFs) have all been implicated in aPL-mediated activation of endothelial cells [66–68], demonstrating again ways in which aPL may co-opt pathways normally associated with more “authentic” inflammatory stimuli.
How does a primed endothelium contribute to thrombosis in patients? Most experimental models of APS trigger thrombosis by explicitly damaging endothelium using laser, ferric chloride, or pinch injury. In these cases studies looking at aPL/endothelium interplay should be interpreted with caution. The model of LPS-priming of the mesenteric microvasculature [44], a possible model of catastrophic APS, circumvents this issue and supports the concept that endothelial activation can trigger simultaneous and widespread thrombosis, at least in the microvascular compartment. However, one must remember that many clinical events are venous and highly localized, suggesting a confined breakdown of normal antithrombotic synergy between endothelium and blood. Experimental models that rely on more authentic thrombotic stimuli, and which characterize endothelium separately from the circulating cell compartment, will be required to resolve these questions.
Monocytes
Monocytes are likely to be important expressers of TF, especially in growing venous thrombi [69]. They are key players in the transition from innate to adaptive immune responses. Circulating monocytes have not been specifically analyzed in animal studies, beyond a recent manuscript stating that introduction of a NOX2 (NADPH oxidase) mutation into the circulating cell compartment (which includes monocytes and excludes endothelial cells) protects against venous thrombosis [53]. In that same study, an antibody targeting TF was also protective.
The ease of access to monocytes has led to their characterization beyond anything done with endothelial cells. For example, lupus patients with aPL have higher monocyte TF production than do lupus patients without, a fact known for 20 years [70], as do primary APS patients with thrombosis [71–73]. Monocytes from APS patients express higher levels of the proangiogenic cytokine VEGF and its receptor Flt-1 [74]. Gene profiling demonstrates upregulation of proinflammatory genes, including TLR8, CD14, and genes associated with oxidative stress [75, 76]. Highlighting the sometimes blurry intersection between coagulation and inflammation, APS monocytes upregulate certain protease-activated receptors (PARs) [77]. Several studies also demonstrate increased levels of monocyte-derived microparticles, a possible important source of TF [63], in circulation [78, 79]. In vitro, aPL trigger monocytes to express TF [80–83], and possibly other proinflammatory cytokines, such as TNF-α and IL-1β [68, 84, 85]. Going forward, it will be interesting to explore the role of monocytes in experimental models with intravital microscopy.
Neutrophils
Neutrophils have recently received attention as key perpetuators of arterial [86, 87], venous [69, 88], and microvascular thrombosis [89, 90]. Relevant to APS, mouse monoclonal aβ2GPI activates neutrophils to release granules and produce hydrogen peroxide [91]. Neutrophils are activated in vitro by human monoclonal aPL [92] and by patient IgG [93, 94], with measurement endpoints that include expression of TF (similar to monocytes) [94], production of IL-8 [92], and release of prothrombotic neutrophil extracellular traps (NETs) [93]. While no consensus exists as to the signaling pathways that lead to neutrophil activation, roles have been suggested for surface β2GPI [91], complement C5a [94], and TLR4 [92, 93]. Some [91], but not all [93], studies suggest a role for the Fc region of IgG. Only recently has attention turned to characterization of neutrophils from APS patients. Similar to monocytes, APS neutrophils display altered mitochondrial membrane potential and evidence of oxidative stress, such as decreased intracellular glutathione [95]. Several groups are interested in the role of NETs in APS [93, 96, 97]. In the thrombosis field, NETs infiltrate arterial and venous thrombi [69, 86–88] and circulate at elevated levels in patients with microthrombosis [89, 90]. Neutrophil extracellular traps are tangles of chromatin and antimicrobial proteins extruded from neutrophils in response to both inflammation and infection [98]. Neutrophil extracellular traps activate platelets and the coagulation cascade and serve as scaffolding upon which a thrombus can assemble [99]; administration of DNase, which disassembles NETs, is protective in animal models of both arterial and venous thrombosis [100, 101]. Like lupus patients, those with primary APS have impaired ability to degrade NETs [96], and APS neutrophils have a lower threshold for NET release [93]. Like lupus patients, APS patients have elevated levels of circulating low-density granulocytes (LDGs) [102]; LDGs represent a subpopulation of neutrophils, of unknown origin, that release NETs in exaggerated fashion [97]. Neutrophils and NETs have only been characterized in one experiment model of APS [54]. Further studies will be needed to understand whether they play a role as initiators or at least perpetuators of the APS prothrombotic phenotype.
Target Receptors and Intracellular Signal Transduction
The activation of target cells by aPL is believed to occur through the interaction of aPL with the main antigen β2GPI, often in dimeric form, and the binding of β2GPI/aβ2GPI complexes to cell surface and intracellular receptors [44]. The molecular mechanisms that lead to aPL-mediated activation of cell types important for thrombosis are summarized in Table 5.2. Many studies report the importance of aPL interaction with cell surface toll-like receptor 4 (TLR4) and annexin A2 (AnnA2) in the activation of ECs in APS [40, 103, 104]. Apolipoprotein endothelial receptor 2 (ApoER2) has also been implicated as a major target for engagement with β2GPI/aβ2GPI complexes on endothelial cells, platelets, and monocytes, most recently as a necessary factor for eNOS inhibition and nitric oxide (NO) downregulation by aPL [41, 49, 105]. Other putative cell surface receptors involved in aPL-mediated EC activation include TLR2, calreticulin, and nucleolin [103, 106].
Table 5.2
Molecular targets and signaling mechanisms in activated cells central to antiphospholipid-mediated thrombosis
Cell type | Receptors | Signaling pathways | Comments |
|---|---|---|---|
Endothelial cell | Main: TLR4, AnnA2, ApoER2 Others: TLR2, calreticulin, nucleolin, complement receptors | p38 MAPK mTOR Ras-ERK NFkB PP2A/eNOS | Multicomponent protein receptor complex formation important in the initiation of cell signal. Activation results in intimal hyperplasia and production of proinflammatory mediators including NO, IL1, IL6, E-sel, ICAM-1, VCAM-1 |
Monocyte | Main: AnnA2, TLR4, TLR2(1/6) Others: TLR7&8, CD14, clathrin | p38 MAPK MEK-1/ERK NFkB/Rel NLRP3 NOX2 c-Jun/AP-1 | Receptor complex formation in lipid rafts important for cell signaling. AnnA2 and TLR4 act in concert. TLR2 mediates aPL uptake into cell. Activation results in production of proinflammatory mediators, primarily TF |
Platelet | GpIbα of GPIb-V-IX, ApoER2 Others: TLR2 | p38 MAPK ERK-1/2 PI3K/Akt | Both GpIbα and ApoER2 necessary for signaling. Activation results in TXB2 production and platelet aggregation. PF4 important for the spatial orientation of aPL when binding to platelet surface. Recent evidence suggests that platelet may be primary aPL target within the microvasculature |
Neutrophil | Main: NETs, PSGL-1 Others: C receptors, TLR4, Fc-γ | MyD88? IFN? | Limited data available. Recent evidence suggests major role for aPL-induced NETs in inducing platelet activation and coagulation. PSGL-1 can accelerate aPL-mediated thrombosis. Proinflammatory phenotype in APS characterized by IFN-related genes, TLR signaling, and Fc-γ activation |
The activation of the p38 MAPK pathway and subsequent nuclear translocation and activation of NFκB is a primary molecular mechanism for aPL activation of ECs [66, 107–109]. Nuclear factor kappa B is a cytoplasmic transcription factor complex that integrates inflammatory signals originating from activated pattern recognition receptors (interleukin-1 receptor/TLRs) and death domain-containing superfamily of cytokine receptors. Nuclear factor kappa B enters the nucleus upon stimulation where it induces the coordinated expression of approximately 5000 genes, the most rapidly inducible genes being those involved in intercellular inflammation, like Gro, IL-8, and IL-6, that may participate in thrombosis in APS [110, 111]. The involvement of the Ras-extracellular signal-regulated kinase (Ras-ERK) pathway in aPL-mediated endothelial cell activation was demonstrated using rat endothelial cells; however, the specificity of this activity to aPL is not confirmed as similar findings were obtained for cells stimulated by LPS and thrombin [112]. The phosphorylation of eNOS S1179 by protein phosphatase 2A (PP2A) is attenuated through aPL interaction with β2GPI and ApoER2 [49]. The mTOR pathway plays a role in vascular stenosis resulting from mechanical endothelial injury; it regulates cellular growth, proliferation, and survival through integration of a variety of signaling proteins. The development of vascular lesions in APS has been recently linked to activation of the mTOR pathway in response to aPL [61].
Annexin A2 and TLR4 are major cell surface targets for aPL-induced activation of monocytes in both venous and arterial thrombosis in APS [68], as has been the engagement of cell surface TLR2, in association with TLR1 and TLR6. Internalization of aPL in monocytes seems to occur via a clathrin- and CD14-dependent process [113]. Most studies indicate that activation of monocytes by aPL induces signaling in the p38 MAPK pathway, with simultaneous and independent activation of the MEK-1/ERK pathway, resulting in nuclear translocation and activation of NFκB/Rel proteins and proinflammatory gene activation [73, 74, 114]. A recent interesting series of experiments provides evidence that intracellular receptors TLR7 and TLR8 are targets of aPL in monocytes [53, 85]. These studies indicate that internalization of cofactor-independent aPL results in increased NOX2 and activation of the NLRP3 inflammasome and is important for inducing venous thrombosis in an APS mouse model. Another recent study shows that activation of the c-Jun/AP-1 pathway may also be important for aPL-induced arterial thrombosis [115].
On platelets, the main receptors that bind β2GPI/aβ2GPI complexes and induce platelet activation include ApoER2 and the glycoprotein Ibα (GPIbα) subunit of the GPIb-V-IX receptor [105, 116]. The p38 MAPK pathway is also important in aPL-mediated platelet activation; potential secondary roles of the ERK-1, ERK-2, and phosphatidylinositol 3-kinase/Akt (PI3K/Akt) pathways have also been suggested [117–119]. Indeed, a mechanism of aPL-induced platelet activation involving the engagement of cell surface GPIbα and TLR2 and the subsequent action of PI3Kβ and α isoforms has recently been described [120]. Platelet factor 4 (PF4), a CXC chemokine secreted and bound by platelets, may also play an important role in platelet activation by stabilizing dimeric β2GPI and facilitating its binding to aβ2GPI and exposed phospholipids and receptors on the platelet surface [117].
Focused research on the role of neutrophils in APS has recently intensified; thus there are limited data on putative receptors and intracellular signaling pathways in APS. Most evidence suggests a major role for NETs in aPL-mediated thrombosis; in fact, DNAse treatment limits thrombus development in APS models [54].
Coagulation Pathways in Antiphospholipid Antibody-Mediated Thrombosis
Much evidence shows that aPL affect hemostasis at multiple levels. Potential mechanisms include activation of platelets, endothelium, monocytes and neutrophils, upregulation of coagulation, downregulation of fibrinolysis, and reciprocal activation of the complement and coagulation systems (Fig. 5.2). However, information is limited regarding the clinical importance and predictive value of these hemostatic changes.
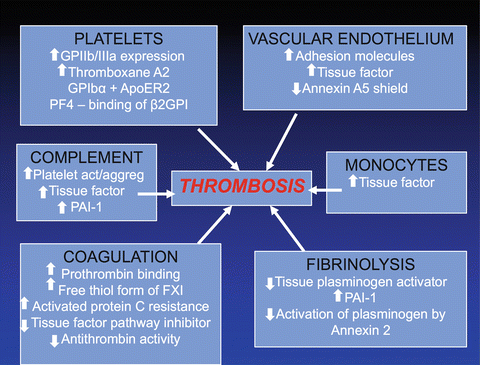
Fig. 5.2
Hemostatic abnormalities in antiphospholipid syndrome
Generation or exposure of TF at the site of trauma is the primary event that initiates clotting. Tissue factor functions as a cofactor for factor VIIa activation of factor X directly and indirectly via activation of factor IX. These coagulation interactions occur on negatively charged procoagulant phospholipids (mainly phosphatidylserine) on the platelet membrane, normally concealed on the inner platelet membrane and translocated in the intact platelet to the outside by membrane “flip-flop” [121]. Tissue factor pathway inhibitor (TFPI) is an important inhibitor of initiation of coagulation. Activation of the TF pathway is integral in the hypercoagulable state of APS, with upregulation of the TF pathway of coagulation [81, 122, 123], to which downregulation of TFPI is contributory [123–125]. Anti-β2GPI suppresses TFPI-dependent inhibition of TF pathway coagulation [124].
The anticoagulant protein C pathway , which plays a central role in regulation of coagulation and maintenance of the fluidity of blood, may also be a key target for aPL. The physiological proteolytic activation of protein C by thrombin occurs on the vascular endothelium and involves two membrane receptors, thrombomodulin and endothelial protein C receptor (EPCR). Binding of thrombin to thrombomodulin shields the procoagulant exosite one of thrombin and facilitates protein C activation [126]. This reaction is intensified by the action of protein C on the endothelial surface by binding to EPCR. Protein C, along with its cofactor protein S, exerts its anticoagulant effect by proteolytic inactivation of phospholipid-bound activated factor V, followed by inactivation of FVIII [127, 128]. Factor Va increases prothrombinase activity by approximately 10,000-fold, and its inactivation by activated protein C (APC) effectively prevents thrombin formation [126]. Although the most common antigenic targets of aPL are the phospholipid-binding proteins β2GPI and prothrombin, protein C and protein S may also be important targets [129, 130]. In vitro aPL effects on the protein C pathway include inhibition of thrombomodulin-mediated activation and anticoagulant activity of APC [130]. Furthermore, aβ2GPI binding to protein C can modulate its action [131] and its subsequent binding to phospholipid surfaces, thereby increasing thrombotic risk [132]. One mechanism proposed to explain how aPL initiate thrombosis is interference with APC’s anticoagulant activity, resulting in acquired APC resistance (APCr) [133]. Thrombotic APS patients have greater APCr to both recombinant human APC and activation of endogenous protein C by ProTac suggesting that high-avidity anti-protein C antibodies, associated with greater APCr , may provide a marker for a severe thrombotic phenotype [134].
Complement activation has a pathogenic role in thrombotic APS [33, 135]. Limited in vitro data suggest that thrombin, factor Xa, and other serine proteases activate complement factors C3 and C5 to C3a and C5a, respectively, producing SC5b-9, the terminal complement component [136]. Complement activation reciprocally amplifies coagulation and inhibits fibrinolysis, through C5a, inducing expression of TF and plasminogen activator inhibitor 1 (PAI-1) [136]. Heparin has an anticomplement effect [137] through its inhibitory effect on thrombin and factor Xa, which cause activation of C3 and C5. Complement activation in APS may be modulated by rivaroxaban, a direct factor Xa inhibitor [138, 139], based on recent evidence that the activation markers C3a, C5a, SC5b-9, and Bb fragment were elevated in warfarin-anticoagulated thrombotic APS patients in the Rivaroxaban in APS (RAPS) trial at baseline [140]. However, C3a, C5a, and SC5b-9 (not Bb fragment) levels decreased in those who switched from warfarin to rivaroxaban, indicating that APS patients with previous VTE, on warfarin, have increased complement activation, likely via the classical pathway, that is decreased by rivaroxaban . Rivaroxaban may therefore provide benefit beyond anticoagulation by limiting complement activation [141].
The Role of Inflammation in Antiphospholipid Antibody-Mediated Thrombosis
Inflammation is rapidly mobilized in response to a variety of foreign and host-derived stimuli. Definitively distinguishing inflammatory response from coagulation is not always straightforward, given the multiple functions of key molecular and cellular components. For example, factor XII, the classic trigger of the contact activation pathway of coagulation, plays an important role in activating inflammatory mediators like bradykinin [142]. Cellular actors like platelets also have multiple roles, from amplification of hemostatic pathways to sensing danger through innate receptors [143]. Investigators have increasingly turned attention to inflammation, as the field attempts to understand how APS targets diverse vascular beds and how those beds rapidly transition from clinically dormant to thrombosed. While alternative anticoagulants will be explored for therapeutic potential, breakthroughs in treatment will likely require understanding of the underlying inflammation.
β2-Glycoprotein-I and Inflammation
β2-Glycoprotein-I is a 326-amino acid glycoprotein, abundant in plasma, that can be deleted from both humans and mice without inducing an obvious phenotype [9]. Initially characterized as a lipid-binding protein [144] and noted to have five complement control protein (CCP) domains reminiscent of the complement regulator factor H [145], β2GPI is now thought to play a role in the innate immune system. Via its domain V, which is enriched with positively charged amino acids, β2GPI binds negatively charged phospholipids such as phosphatidylserine. Given that such phospholipids are exposed on the cell surface as an “eat me” signal during apoptosis, investigators speculate that β2GPI facilitates clearance of apoptotic cells. Indeed, β2GPI binds to phosphatidylserine-expressing liposomes and cells in vitro [146], serving as a bridge to phagocytes [147]. Further, phosphatidylserine-containing liposomes are cleared more efficiently in mice when bound to β2GPI [148]. While the specific receptors involved in clearance are not known, they are likely of the LDL receptor family [149].
The concept of β2GPI as scavenger applies in other situations. β2-Glycoprotein-I can partner with factor H to downregulate the alternative complement pathway [150]. Recent work shows that β2GPI binds bacterial LPS to promote its neutralization and clearance [149]. β2-Glycoprotein-I also functions as a sink for oxidative stress through its free thiol groups, with the oxidized form of β2GPI (lacking free thiols) detected at high levels in APS patients compared to healthy subjects, other autoimmune disease patients, and thrombotic disease controls [151, 152]. Krilis et al. propose that oxidized-β2GPI levels may serve as a biomarker of thrombotic risk, and they have developed an ELISA to measure posttranslational redox modifications of β2GPI, including total β2GPI and free thiol-β2GPI. They also hypothesize that free thiol-β2GPI format is protective in APS because thiol groups prevent hydrogen peroxide-induced cell injury; a decrease in free thiol groups via oxidation increases risk for oxidative stress-induced injury [153]. This topic is discussed in greater detail in Chap. 2.
β2-Glycoprotein-I may play an active role in combatting infections, as neutrophil proteases cleave the full-length protein to generate antimicrobial cationic peptides [154]. β2-Glycoprotein-I deficiency does not seem to be profoundly immunosuppressive, and the β2GPI function(s) most important for host defense remain open to debate. Nevertheless, this key APS antigen engages with the immune system and inflammation in multiple ways, setting the stage for a break in tolerance that leads to autoimmunity.
Complement System
“Complement ” describes a system of circulating proteins that impact and activate each other via proteases, thereby promoting inflammatory cell recruitment, opsonization with pathogen clearance, and cell death. Often described as a cascade, the system can be activated by different stimuli with eventual convergence at the level of C5a generation (a chemotactic and proinflammatory protein) and assembly of the membrane attack complex (via components C5b, C6, C7, C8, and C9) [155]. The complement system plays a role in systemic lupus as a mediator of tissue damage [156].
The most compelling evidence implicating complement in APS comes from animal models. After early work showed that antagonizing complement protects against pregnancy loss [157], attention turned to complement’s potential role in aPL-mediated thrombosis. In the femoral vein pinch injury model, targeting complement C3, C5, and C6 are all protective [33, 37, 39, 42]. Similarly, antagonizing C5 or C6 is protective in the LPS/mesenteric circulation model [44].
Given the antibody dependence of APS, it is tempting to speculate that activation of the complement system by the classical pathway (which is initiated by antibodies) is central to pathogenesis; however, F(ab’)2 fragments or artificial β2GPI dimers alone activate thrombosis in various models, arguing that the alternative pathway (spontaneous activation that gets amplified by pathogens or tissue damage that is not dependent on antibodies) may be the more important player [41, 43, 47]. This idea is supported by evidence of low-grade complement activation in APS [158–160] via the alternative pathway [161–163]. On the contrary, there are studies pointing to an association between aPL and classical pathway activation [135, 164, 165]. It is conceivable that aPL, β2GPI (a regulator of factor H), and their complexes drive activation through both pathways. While the method by which complement activation might promote thrombosis is unclear, both vascular damage (via the membrane attack complex) and leukocyte recruitment (via C5a) are possibilities. A reason to probe these pathways is that complement inhibitors are likely to be increasingly used in clinical practice, with an example in APS-associated thrombosis being recently described [166].
Toll-like Receptors (TLRs)
Toll-like receptors evolved to recognize non-host molecular patterns that characterize pathogen invasion, the classic example being the recognition of LPS by surface-expressed TLR4. Displayed by leukocytes, TLRs rapidly trigger proinflammatory cytokine release and cell activation, classic examples of innate immune function [167]. Toll-like receptor 4 is the only family member to be studied extensively in mouse models of APS, where its deletion protects against venous and arterial thrombosis [27, 51, 52]. Work with patient samples is limited; recent work suggests that APS monocytes are primed to express TLR2 and TLR4 on their surface when stimulated [168], while APS dendritic cells may overexpress endosomal TLR7 [169].
In vitro studies have probed the role of TLRs in mediating cell activation by aPL, especially aβ2GPI, the rationale being the observation that some cell surface receptors for β2GPI, such as annexin A2, do not have a cytoplasmic tail to mediate signaling. MyD88, an adapter protein that conveys signals from almost all surface TLRs (including TLR2 and TLR4), was identified more than a decade ago as a factor in aβ2GPI-mediated activation of endothelial cells [170]. Subsequent studies implicated TLR4 as an aPL co-receptor on endothelial cells, including one that found a role for a complex consisting of annexin A2, TLR4, calreticulin, and nucleolin [103]. Toll-like receptor 4 has a role in the in vitro activation of monocytes [68, 84, 171, 172] and neutrophils [92, 93] by aPL. Whether LPS itself is a player in these pathways, either as a stimulator of TLR expression [92, 173, 174] or as a bridge between β2GPI and TLRs [175, 176] is an area of ongoing investigation.
Some studies point to TLR2 as an aPL co-receptor [113], possibly excluding a role for TLR4 [106, 173]. Endosomal TLRs like TLR7 and TLR8 seem to be primed for hyperresponsiveness by aPL [169], creating a situation in which aPL amplifies production of proinflammatory cytokines such as IL-1β [85] or interferons. Because the vast majority of studies use in vitro systems and different types of aPL, it is difficult to create a cohesive model. Interestingly, β2GPI depletion in male BXSB-Yaa mice, a mouse model of SLE dependent on duplication of TLR7, accelerates and potentiates the autoimmune phenotype implying that the main antigen in APS, β2GPI, has a regulatory effect on TLR7 [177]. On balance the evidence supports a role for TLRs in APS, as mediators of cell activation and as key signaling molecules, which could tip an aPL-primed system toward thrombosis in response to environmental triggers.
What Is Controversial and/or Unknown?
Distinct Structural and Functional Characteristics of Antiphospholipid Antibody Types Related to Thrombosis
Experimental evidence implicates pathogenic aPL-induced activation of diverse cell surface receptors and intracellular pathways to promote thrombosis. This section will review whether the thrombogenic effects of these aPL can be distinguished by isotype, binding properties, and/or functional effects upon target cells.
Structural Characteristics
Strong evidence for pathogenicity relates to IgG binding the N-terminal domain (domain I or DI) of β2GPI [36, 178], simultaneously cross-linking two β2GPI molecules, activating biological pathways. There is emerging interest on the importance of other isotypes, particularly IgA aPL, in the pathogenesis of APS.
β2-Glycoprotein-I contains five domains (DI-DV) and anchors to anionic PL via DV. Although antibodies directed against all domains have been reported, IgG anti-DI are most closely linked to APS and are elevated in patients with APS compared to disease and healthy controls [178–184]. Both affinity-purified IgG anti-DI from APS serum [29] and a human monoclonal anti-DI IgG aPL [10, 185] are prothrombotic in mice [186, 187]. In the same mouse model, recombinant human DI abrogates aPL-induced thrombosis [36]. A human monoclonal IgG anti-DI is, in two different animal models, prothrombotic and capable of causing fetal loss in naïve mice treated with LPS [45].
The significance of IgM aPL against DI is unclear; de Laat and colleagues reported that IgM anti-DI had no increase in their association with venous thrombosis compared to IgM aβ2GPI [181]. Two studies reported that more than 50% of patients with IgA aβ2GPI had reactivity against DIV-V of β2GPI [188, 189]. Pericleous et al. [190] compared IgG, IgM and IgA aCL, and aβ2GPI and anti-DI in patients with APS, SLE (no APS), and healthy controls; IgG aPL was the most common and highest-titer aPL, while IgA aβ2GPI and anti-DI correlated more strongly with APS than did IgM. They also found that inclusion of IgG, IgM, or IgA anti-DI positivity increased the odds for APS detection compared to aCL and/or aβ2GPI positivity alone; while IgG aCL, aβ2GPI, anti-DI, and IgA anti-DI were associated with thrombotic but not obstetric complications.
Immunoglobulin A aCL are found in patients with SLE, many of whom do not have APS [191]. In contrast, isolated IgA aβ2GPI positivity (in patients negative for IgG/IgM-aCL/aβ2GPI and LA) is associated with thrombotic and obstetric manifestations of APS [192], and IgA aβ2GPI are prothrombotic in mice [188]. For these reasons, published reviews propose testing for IgA (particularly aβ2GPI) aPL only in IgG/IgM-aCL/aβ2GPI and LA negative patients in whom APS is strongly suspected [193]. Readers should be aware of ongoing efforts to update laboratory and clinical classification criteria for APS highlighted at the recent 15th International Congress on Antiphospholipid Antibodies (discussed in Chap. 15).
Functional Characteristics
Cell surface receptors that interact with aPL and/or β2GPI include annexin A2, ApoER2, and TLRs. Intracellular signaling through MAPK and the transcription regulator NFκB is demonstrable in aPL-mediated activation of target cells and is directly linked with TLR activation. Poulton et al. [194], systematically analyzed original studies on the effects of aPL on cell surface receptors and cell signaling pathways and found evidence, from multiple approaches, that TLR4, p38 MAPK, and NFκB mediate pathogenic effects of aPL in thrombotic APS.
Heterogeneity among studies may derive from the fact that aPL in patients with thrombosis has different properties from aPL in patients with no thrombosis (obstetric APS). Few studies have compared effects on target cells of aPL from patients with and without thrombosis. In a series of papers, Lopez-Pedrera et al. [73, 74, 77] examined monocytes from patients with APS. They found differences in monocyte expression of p38 MAPK, NFκB signaling pathways, TF, VEGF, soluble Flt-1, and PAR1 and 2 in APS patients with thrombosis compared to APS patients with no thrombosis. Lambrianides et al. [171] found that IgG from these two APS patient clinical groups had different effects on p38 MAPK and NFκB activation in monocytes.
Lopez-Pedrera et al. [195] used traditional proteomics techniques to quantify 22 proteins in monocytes of 51 patients with APS (32 with thrombosis and 19 with pregnancy morbidity alone) and controls. Six proteins (annexin I, annexin II, protein disulfide isomerase, Nedd8, RhoA proteins, and Hsp60) most significantly altered, from monocytes of patients with thrombotic APS, were functionally related to induction of a procoagulant state and to autoimmune responses. They were subsequently found to be regulated by statin therapy [196]. Proteins implicated in recurrent spontaneous fetal loss such as fibrinogen and hemoglobin were also dysregulated in patients with obstetric APS.
More recently, Ripoll-Nunez et al. [197] used new proteomics techniques to analyze monocytes treated with IgG from 27 patients with different manifestations of APS. They found that four of the most significantly regulated proteins—vimentin, zinc finger CCH domain-containing protein 18, CAP Gly domain-containing linker protein 2, and myeloperoxidase—were differentially regulated in monocytes treated with thrombotic or obstetric APS IgG compared with healthy control IgG. They further characterized the proteome of thrombotic APS IgG-treated monocytes and found that many proteins identified possessed immune response, cytoskeletal, coagulation, and signal transduction functions relevant to APS. No single pathway is known to be dominant at this time.
Other studies demonstrate evidence for cytoskeletal protein involvement in endothelial activation in APS. Betapudi et al. [198] found that aβ2GPI mediated induction of endothelial microparticle release depends on phosphorylation of the myosin regulatory light chain and assembly of actin-myosin networks. In addition, involvement of cytoskeletal proteins in aβ2GPI-mediated activation of EC (via interactions with surface receptor annexin A2, which lacks a transmembrane domain) may explain how engagement of this receptor leads to activation of intracellular signaling pathways. Allen et al. [103] demonstrated that signaling through TLR4 is mediated through assembly of a multi-protein (annexin A2, TLR4, calreticulin, and nucleolin) signaling complex on the EC surface, so cytoskeletal protein may contribute to aPL-mediated EC activation and thus thrombosis.
Other studies show that aPL promote thrombosis by disruption of homeostatic and cytoprotective signaling in endothelial cells. An association between increased activity of the pro-survival mTOR pathway and endothelial hyperplasia in renal biopsies has been reported in patients with APS compared to patients with lupus nephritis [61]. Other studies demonstrate aPL-mediated dysregulation of endothelial nitric oxide synthase (eNOS), which is responsible for maintenance of a healthy endothelium. Ramesh et al. [49] found that thrombotic-APS IgG inhibits eNOS activity in cultured human EC treated with VEGF, leading to inflammation and oxidation. Similarly, Ulrich et al. [199] found that the ability of thrombotic-APS IgG to antagonize in vivo and in vitro endothelial repair is mediated by eNOS deficiency.
Ramesh et al. [49] and Ulrich et al. [199], using ApoER2 null (−/−) mice, demonstrated a loss of APS IgG-mediated effects, thus implicating ApoER2 in aPL-mediated EC activation. Romay-Penebad et al. [41] directly studied involvement of this receptor in aPL-mediated thrombosis and found that the thrombogenic effects of a single IgG APS and of a constructed dimeric form of β2GPI (that in vitro mimics β2GPI-antibody immune complexes) were reduced in ApoER2 (−/−) compared to wild-type mice. Those effects induced by IgG APS and by the dimer were reduced by treatment of wild-type mice with soluble binding domain 1 of ApoER2 (sBD1), which inhibits binding of dimerized β2GPI to ApoER2. Therefore, ApoER2 may also be involved in pathogenesis of APS thrombosis. This effect may not be specific to overt thrombosis since ApoER2 is required for the adverse effect of aPL on pregnancy outcomes in mice [200].
Related posts:
 Recent Advances in Understanding of the Genetics of Antiphospholipid Syndrome
Clinical and Prognostic Significance of Non-criteria Antiphospholipid Antibody Tests
Prevention and Treatment of Obstetric Antiphospholipid Syndrome
15th International Congress on Antiphospholipid Antibodies Task Force on Catastrophic Antiphospholipid Syndrome Report
Recent Advances in Understanding of the Genetics of Antiphospholipid Syndrome
Clinical and Prognostic Significance of Non-criteria Antiphospholipid Antibody Tests
Prevention and Treatment of Obstetric Antiphospholipid Syndrome
15th International Congress on Antiphospholipid Antibodies Task Force on Catastrophic Antiphospholipid Syndrome Report
 Mechanisms of Antiphospholipid Antibody-Mediated Pregnancy Morbidity
Mechanisms of Antiphospholipid Antibody-Mediated Pregnancy Morbidity
 Treatment of Non-criteria Manifestations in Antiphospholipid Syndrome
Treatment of Non-criteria Manifestations in Antiphospholipid Syndrome
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


