Fig. 6.1
Blocking the actions of antiphospholipid antibodies (aPL). (A) The monoclonal antibody, N11, prevents the actions of aPL on trophoblast and endothelial cells and attenuates aPL-associated pregnancy complications and thrombosis in mice. (B) The synthetic peptide TIF1 is a competitive blocker that displaces β2GPI from the surface of the trophoblast and endothelium, thus preventing aPL binding and aPL-mediated thrombosis and pregnancy complications in vivo. (C) Eculizumab inhibits C5 cleavage and prevents pregnancy loss in experimental models. (D) The human monoclonal, MBB2ΔCH2, binds domain I of β2GPI but fails to activate complement. MBB2ΔCH2 competes with patient aPL and prevents their pathogenic effect
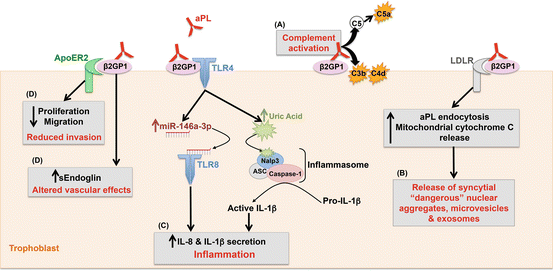
Fig. 6.2
Effect of antiphospholipid antibodies (aPL) on trophoblast cells. aPL recognizing β 2 GPI expressed by the trophoblast: (A) activates complement on the cell surface; (B) become internalized via low-density lipoprotein receptor (LDLR) family members and in turn promote the deportation of “dangerous” syncytial nuclear aggregates and other microvesicles; (C) triggers secretion of inflammatory cytokines and chemokines by activating TLR and inflammasome pathways; and (D) promotes an anti-angiogenic profile and through ApoER2 reduces cell proliferation and migration
Because activated complement fragments have the capacity to bind and damage self-tissues, autologous bystander cells must be protected. To this end, most human and murine cells express soluble and membrane-bound molecules that limit the activation of various complement components at sited of inflammation [23]. Though activated complement components are present in normal placentas [24, 25], it appears that in successful pregnancy, uncontrolled complement activation is prevented by three regulatory proteins present on the trophoblast membrane: decay accelerating factor (DAF), membrane cofactor protein (MCP), and CD59 [26, 27]. All three proteins are strategically positioned on the trophoblast, along with circulating soluble regulators, and provide a mechanism to protect the fetus from damage due to activation of the complement pathway. Intact complement regulation is essential for maintenance of normal pregnancies, because in pregnant mice deficient in cell-bound regulators of complement activation, fetuses die in utero surrounded by inflammatory cells and complement split products; breeding mice that lack complement inhibitors on a complement-deficient background rescues pregnancies [28, 29].
As discussed below, phosphatidylserine, externalized during trophoblast differentiation , allows β2GPI to be expressed on the cell surface and, therefore, provides a target for aPL [30, 31], which can activate complement via the classical pathway to generate split products that mediate placental injury and cause fetal loss and growth restriction. The exaggerated complement activation that results may overwhelm the inhibitory capacity of local complement regulatory proteins allowing the complement cascade to proceed.
Studies in women support the role of complement in aPL-associated pregnancy complications. C4d is present in placentae from women with SLE and/or APS and from women with preeclampsia [32–34]. Indeed, in a systemic review literature of histology in placentae, one of the most common features in the placentae of aPL-positive women compared to control women was deposition of complement split product C4d [35] (Fig. 6.2A). Furthermore, in a prospective observational study in pregnant SLE/aPL patients , elevated levels of the alternative pathway complement activation product Bb are detectable early in pregnancy and are strongly associated with adverse pregnancy outcomes [36]. Mild hypocomplementemia has also been reported in primary APS in two studies [37, 38].
Observational studies in aPL pregnancies also emphasize the importance of dysregulation of the complement system in adverse pregnancy outcomes [39, 40]. Dysregulation can present as either excessive activation or inadequate regulation of this complex system. Soluble and membrane-bound complement regulatory proteins protect by limiting spontaneous alternative pathway activation. Indeed, defective function of complement regulators is associated with inflammatory and thrombotic injury associated with hemolytic uremic syndrome and glomerulonephritis [41, 42]. A report that 18% of 40 patients with SLE and/or aPL who had preeclampsia had heterozygous mutations in genes encoding three complement regulatory protein-membrane cofactor protein (MCP), complement factor I, some previously identified in atypical hemolytic uremic syndrome, a disease characterized by endothelial damage links complement activation to disease pathogenesis [43], underscores the role of complement in obstetric APS.
Inflammatory Mediators Downstream of Complement Activation as Effectors of Fetal Damage in Experimental Models of Antiphospholipid Syndrome
There are multiple effectors of fetal injury downstream of complement activation, triggered by C5a–C5a receptor interactions. Tumor necrosis factor (TNF)-α is one mediator that links complement activation and pathogenic Antiphospholipid antibodies to fetal damage. Antiphospholipid antibodies, specifically targeted to decidual tissue, cause a rapid increase in decidual and systemic TNF-α levels. Complement 5-deficient mice were protected from fetal death and showed no increase in TNF-α levels identifying TNF-α as a critical intermediate that acts downstream of C5 activation [13]. That TNF-α is itself pathogenic is suggested by studies showing that miscarriage induced by aPL is less frequent in mice deficient in TNF-α or treated with TNF-α blockade [13]. Furthermore, in antibody-independent models of placental insufficiency and preeclampsia, complement activation at the maternal-fetal interface leads to elevation in local TNF-α levels, reduction of the essential angiogenic factor, vascular endothelial growth factor (VEGF), and, ultimately, abnormal placentation and fetal death. Blockade of complement activation or blockade of TNF-α improves spiral artery remodeling and rescues pregnancies, underscoring the relationship of these mediators and their importance [44].
Complement 5a also triggers fetal damage through induction of tissue factor (TF) expression. Treatment with aPL increases TF in neutrophils which enhances oxidative burst providing a mechanism for trophoblast injury and pregnancy loss triggered by these autoantibodies [19, 45]. Finally, complement activation products may cause an imbalance of angiogenic factors required for normal pregnancy. Satisfactory development of the fetomaternal vasculature is required for successful embryonic growth, and insufficient placental vascularization has been associated with early embryonic mortality, preeclampsia, and IUGR [46]. Normal placental development requires coordinated expression of angiogenic growth factors, and C5a–C5a receptor interactions trigger release of anti-angiogenic factors from leukocytes which can alter the balance of angiogenic factors in pregnancy and lead to the pregnancy complications associated with APS [16].
Antiphospholipid Antibody-Trophoblast Interactions
The placenta is a major target for aPL, in particular β2GPI-dependent antibodies, which bind to human trophoblast. This may explain why pregnancy complications associated with placental development and function occur in women with APS. Indeed, the expression of β2GPI on the placenta and, in particular, on trophoblast cell membranes is the prerequisite to explain aPL-placental tropism. While most cells will only bind β 2 GPI on their cell surface under pathologic, stimulatory, or apoptotic conditions, when the inner negatively charged phospholipids become exposed onto the outer leaflet of the plasma membrane, the trophoblast is unusual in that it normally expresses these anionic phospholipids on its cell surface. This occurs as a result of the trophoblast’s high level of proliferation and differentiation that is associated with tissue remodeling during placentation [47, 48]. As a result, the positively charged plasma protein, β2GPI, can bind to phosphatidylserine exposed on the external cell membranes of trophoblast undergoing syncytium formation, although additional receptors may also be involved [6, 7, 49]. Furthermore, the trophoblast synthesizes its own β 2 GPI, and this protein translocates to the cell surface [50]. In vivo, there is evidence of β 2 GPI localized to the surface of the extravillous trophoblast cells that invade the decidua and to the syncytiotrophoblast cells that are in direct contact with maternal blood [50, 51]. β 2 -glycoprotein-I binds to the surface of the human trophoblast through the phospholipid-binding site in the fifth domain of the molecule, thus offering suitable epitopes for the maternal autoantibodies [6, 7, 49]. Hence, β2GPI-dependent aPL appear to represent the main pathogenic autoantibodies in obstetrical APS. Accordingly, it has been hypothesized that most of them could be absorbed at the placental level (where β2GPI is expressed) and not transferred to the fetus . This would explain why thrombotic events are rarely reported in babies born to aPL-positive mothers in spite of the high thrombophilic profile of neonates [52]. Although the syncytiotrophoblast and extravillous trophoblast populations both bind aPL recognizing β2GPI, only the syncytiotrophoblast internalizes these antibodies via low-density lipoprotein receptors (LDLR) [53, 54]. The consequence of this aPL internalization is syncytiotrophoblast mitochondrial leak of cytochrome C [53] (Fig. 6.2B).
Since aPL bind to the trophoblast, it seems likely that the pathogenesis of pregnancy failure/complications in patients with APS is initiated at the placenta. Consequently, a number of studies have sought to evaluate the effects of aPL on trophoblast cells in vitro and have found that these autoantibodies affect several cell functions. Studies using human term trophoblast or choriocarcinoma cells show that aPL inhibit proliferation and syncytia formation [55–57], alter adhesion molecule expression [58], reduce invasiveness [56, 59–62], and decrease human chorionic gonadotropin [56, 61]. However, women with APS and pregnancy failure have circulating aPL at the time of implantation, and the most frequent clinical outcome is early pregnancy loss. Thus, more recent studies have shifted their focus to understanding the effects of aPL on the first trimester trophoblast. First trimester placental explants exposed to aPL have also been shown to produce less human chorionic gonadotropin (hCG) [63]. First and third trimester placental explants exposed to aPL generate distinct metabolic markers, ceramide, and diacylglycerols that may be involved in trophoblast responses to aPL [64]. Other studies using first trimester placental explants have found that the serum of patients with SLE/APS and recurrent pregnancy loss, anticoagulant-containing sera, or patient-derived aPL cause increased trophoblast apoptosis [63, 65, 66]. Anti-β2GPI antibodies also augment the non-apoptotic shedding of trophoblastic material from first trimester placental explant cultures [67]. This is an important observation since during normal pregnancy, the placenta constitutively releases trophoblast microparticles, mononuclear trophoblast, and trophoblast syncytial knots from its outer syncytiotrophoblast layer into the maternal circulation [68], and in preeclampsia, a common outcome in APS-complicated pregnancies, shedding, or deportation of this material is significantly increased [69] (Fig. 6.2B).
Using in vitro cultures of human first trimester trophoblast cell lines and primary cells, mouse anti-β2GPI monoclonal antibodies and purified patient-derived polyclonal aPL with β2GPI reactivity have been found to enhance cytokine and chemokine secretion, specifically interleukin (IL)-8, IL-1β, growth-regulated protein (GRO)-α, and monocyte chemotactic protein (MCP)-1, which might explain the immune cell infiltration seen at the maternal-fetal interface. This aPL-induced inflammatory response is mediated by the Toll-like receptor 4 (TLR4)/MyD88 pathway [70], most likely because of molecular mimicry between β2GPI and bacterial components, like lipopolysaccharide (LPS) [71, 72]. Indeed, in vivo studies have shown that animals immunized with microbial components develop aβ2GPI antibodies and APS-like symptoms [73, 74]. Downstream of TLR4, trophoblast secretion of IL-1β secretion is mediated by the induction of endogenous uric acid, which in turn activates the Nod-like receptor (NLR), Nalp3, leading to Nalp3/ASC/caspase-1 inflammasome activation, and subsequent IL-1β [70, 75]. In parallel, IL-8 secretion is mediated by the induction of miR-146a-3p, which in turn activates the RNA sensor, TLR8 [76]. Thus, aPL-induced miR-146a-3p and uric acid act as endogenous secondary signals for trophoblast TLR8 and Nalp3 inflammasome activation, to drive trophoblast inflammation (Fig. 6.2C). The aPL-associated upregulation of trophoblast-derived uric acid and miR-146a-3p was found to be mirrored in the serum of women with aPL and adverse pregnancy outcomes [75, 76].
In parallel to the aPL-induced inflammatory response, aPL diminish the trophoblast’s ability to migrate, independently of the TLR4 signaling pathway, by inhibiting the cell’s constitutive production of IL-6, which in turn leads to decreased STAT3 activity [77], a critical mediator of trophoblast invasiveness [78]. This reduction in trophoblast migration was also found to involve aPL-induced upregulation of TIMP2 [79]. In a follow-up study, the surface-expressed receptor, apolipoprotein E receptor 2 (apoER2), was found to mediate this reduced migration [80]. However a recent study reported that patient-derived aPL reduce trophoblast invasion in a TLR4-depedent manner [81] (Fig. 6.2D).
Lastly, aβ2GPI disrupts the basal trophoblast angiogenic factor balance, by inducing the secretion VEGF, PlGF, and sEndoglin levels (Fig. 6.2D). Although TLR4 is not involved in this response, functional MyD88 was required for the aPL-induced upregulation of PlGF, suggesting that receptors other than TLR4 that utilize MyD88, such as IL-1R or TLR8 [75, 76], are involved. All these aPL-mediated effects may play a role in causing a defective placentation [6, 7, 49]. Indeed, using an in vitro model of spiral artery transformation, aPL and sera from APS patients with pregnancy morbidity were shown to disrupt the normal trophoblast-endothelial cell interactions leading to reduced trophoblast-endothelial tube stability [82].
Data also suggest that aPL can cause abnormalities at the maternal side of the placenta. Impaired endometrial differentiation and lower expression of complement regulatory proteins (DAF/CD55) were found in endometrial biopsies from APS patients. These alterations before conception may compromise implantation and predispose to complement-mediated pregnancy failure [83, 84]. In addition, β2GPI-dependent aPL are able to react with human stromal decidual cells in vitro and induce a pro-inflammatory phenotype [85]. These findings do suggest that APS-associated pregnancy complications can be mediated by several distinct pathogenic events.
At variance with the vascular manifestations of the syndrome, the two-hit hypothesis may not fit well with the APS obstetrical manifestations [6]. Passive infusion of IgG fractions with aPL activity induces fetal loss in naive pregnant mice and does not apparently require a second hit. β2-glycoprotein-I is largely expressed in placental tissues even in physiological conditions [51, 86], and binding of labeled exogenous β2GPI infused into naïve pregnant mice to trophoblast and endothelial in the labyrinth was recently documented in vivo by eXplore Optix™ imager [87]. Thus, the large availability of the target antigen for pathogenic aPL at the placental level is in strong contrast with the lack of a comparable expression in other tissues of naïve mice and even in highly vascularized human tissues such as the kidney [7]. It is possible to speculate that a high expression of β2GPI at the placental level, together with the hormonal and blood flow modifications linked to the pregnancy, is sufficient to favor the pathogenic activity of the autoantibodies without any additional factor.
Antiphospholipid Antibody Internalization into the Syncytiotrophoblast
Preeclampsia, a hypertensive disorder unique to human pregnancy, is one of the obstetric conditions often associated with aPL, and aPL have been reported to increase a woman’s risk for developing preeclampsia almost tenfold [88–95]. While the pathogenic processes leading to preeclampsia are not fully understood, it is apparent that a toxin or toxins released from the placenta trigger maternal endothelial cell dysfunction . This maternal endothelial cell dysfunction is thought to be key to development of the maternal disease and may precede the onset of the clinical signs of preeclampsia by several weeks.
The human placenta is covered by a single multinucleated cell, the syncytiotrophoblast, which is bathed in maternal blood. Most cells produce subcellular lipid enclosed packages called extracellular vesicles that are released into the extracellular environment and are important in intercellular signaling. Extracellular vesicles from mononuclear cells are limited to microvesicles and nano-vesicles, some of which are exosomes. However due to its multinucleated structure, the syncytiotrophoblast also produces a unique type of multinucleated macro-vesicles called syncytial nuclear aggregates (SNAs), which are thought to be the end of the life cycle of the syncytiotrophoblast and therefore may be somewhat equivalent to apoptotic blebs produced by mononuclear cells. These multinucleated vesicles are larger than most mononuclear cells, being on average 72 μm in length and often have a tear-dropped shape [96, 97]. Syncytial nuclear aggregates are extruded from the surface of the syncytiotrophoblast directly into the maternal blood, which deports them to the maternal lungs where they become trapped in the pulmonary capillaries (the site where they were first identified in association with eclampsia over 120 years ago)[98]. While about 100,000 SNAs are deported daily from the placenta in all normal pregnancies [99], there is 20-fold increase in the number of SNAs deported in preeclampsia [96, 100, 101]. Syncytial nuclear aggregates are cleared from the maternal pulmonary capillaries on average in 3–4 days, and there is evidence that this clearance is due to phagocytosis of the SNAs by the maternal pulmonary endothelial cells against which they are trapped [102–104]. It has been suggested that SNAs from preeclamptic placentae may be one of the placental toxins that trigger preeclampsia [104, 105].
It has long been known that aPL can interact with the syncytiotrophoblast since, as described in the preceding section, aPL can reduce production of hCG, reduce formation of syncytiotrophoblast, and have been shown to disrupt the annexin V anticoagulant shield on the surface of the syncytiotrophoblast [10].
More recently it was reported that aPL, when incubated with first trimester placental explants, increases production of syncytial nuclear aggregates from the syncytiotrophoblast [103, 106]. So how do aPL induce the production of SNAs? Using a combination of murine monoclonal and patient-derived aPL, it was shown that aPL rapidly penetrate the syncytiotrophoblast with this process requiring as little as 2 min [53]. The internalization of the antibodies was receptor dependent. While the exact identity of the receptor remains unclear, transport of aPL into the syncytiotrophoblast was inhibited by receptor-associated protein (RAP) , a low-density lipoprotein receptor (LDLR) family-binding protein (and by chloroquine) suggesting that aPL enter the syncytiotrophoblast via LDLR-mediated endocytosis [53] (Fig. 6.2B).
Immunofluor-gold electron microscopy was used to show that once internalized into the syncytiotrophoblast, the aPL were bound to mitochondria and that there was an increase in the number of mitochondria with swollen morphology associated with the aPL [53]. Further, once internalized, aPL disrupted mitochondrial function resulting in decreased oxidative phosphorylation though complex IV and the translocation of cytochrome C from the mitochondria to the cytoplasm of the syncytiotrophoblast [53]. This movement of cytochrome C has the potential to increase cell death in the syncytiotrophoblast by promoting the formation of the apoptosome, while the disruption of mitochondrial function may lead to a more necrotic type of death process in the syncytiotrophoblast. It is likely that, via these effects on the mitochondria, aPL increase the production of SNAs (Fig. 6.2B).
Not only do aPL increase the number of SNAs extruded from the placenta but importantly they change the nature of SNAs. It has been shown that SNAs from normal first trimester placenta, when phagocytosed by endothelial cells, render those endothelial cells resistant to activation, and this is important to the normal maternal adaptation to pregnancy [102]. In stark contrast, the SNAs extruded from placentae exposed to aPL become “dangerous” and induce endothelial cell activation similar to that seen in preeclampsia [104] (Fig. 6.3B). Providing further evidence that aPL induce the syncytiotrophoblast to extrude dangerous SNAs as a result of an interaction with the mitochondria, a proteomic comparison of SNAs from aPL or control antibody-treated placentae showed that of 72 regulated proteins 13 were involved in mitochondrial function [106].
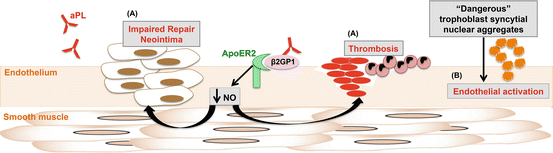
Fig. 6.3
Effect of aPL on the endothelium. (A) aPL-β2GPI interaction with apoER2 impairs the production of nitric oxide (NO) leading to increased leucocyte adhesion, thrombosis, and neointima hyperplasia. (B) The “dangerous” syncytial nuclear aggregates extruded from placentae exposed to aPL induce endothelial cell activation
It is not yet entirely clear what changes aPL induce in SNAs to make them dangerous, but it has been shown that the alarmin, high mobility group box 1 (HMGB1), is increased in the cytoplasm of SNAs from aPL-treated placentae. Similarly, the amount of “free” mitochondrial DNA, which is also an inflammatory danger signal, is increased in the smaller micro- and nano-vesicles that are released from aPL-treated placentae. Thus, aPL rapidly enter the syncytiotrophoblast via a receptor-mediated mechanism and disrupt mitochondrial function, resulting in the release of dangerous SNAs into the maternal blood. Maternal endothelial cells become activated in response to these dangerous SNAs in the same manner as occurs in preeclampsia. This provides a potential mechanism to explain why aPL are such a potent risk factor that predispose women to develop preeclampsia.
The Role of ApoER2 in Antiphospholipid Antibody-Mediated Placental and Endothelial Dysfunction
Apolipoprotein E receptor 2 (apoER2) , also known as LRP8, is a member of the low-density lipoprotein (LDL) receptor family, which includes LDL receptor, LRP1, megalin, and very low-density lipoprotein receptor. When human apoER2 cDNA was first cloned, abundant expression of mRNA of the receptor was detected in the brain and in the placenta [107]. Subsequently, a critical role of the receptor was discovered in neuronal development as a receptor for reelin [108–110]. In the neuronal cells, reelin binding to apoER2 initiates activation of a series of kinases to regulate cellular function required for normal brain development [109–112]. More recently, it has been shown that the receptor is highly expressed in vascular cells, including platelets, monocytes/macrophages, vascular smooth muscle cells, and endothelial cells [113–120]. In 2003 de Groot’s group reported for the first time that that apoER2, a splice variant of apoER2 expressed in platelets , can interact with dimerized β2GPI in platelets [121]. Because aβ2GPI-β2GPI complex plays a key role in pathogenesis of APS, a series of studies ensued in in vitro experiment, in cell culture, and in mouse models to determine the role of apoER2 in the actions of aPL [119, 122–126].
In endothelial cells, our group (Chieko et al.) found that monoclonal aβ2GPI or polyclonal human aPL attenuate activity of endothelial nitric oxide (eNOS) through dephosphorylation of the enzyme at Ser1177, the critical phosphorylation site for the activity of the enzyme, leading to impaired production of nitric oxide (NO) [119] (Fig. 6.3A), which is a key signaling molecule for maintenance of normal vascular function. Nitric oxide produced by eNOS stimulates vascular relaxation, prevents endothelial inflammation, attenuates platelet activation, and reduces smooth muscle cell proliferation and migration [127, 128]. In cultured endothelial cells, we further demonstrated that inhibition of eNOS by aPL contributes to attenuated cell migration and increased adhesion to monocytes and that these aPL actions were abrogated with apoER2 knockdown by siRNA [119]. Mirroring the findings in culture, aPL administration in wild-type mice decreased eNOS-mediated vascular relaxation, impaired carotid artery reendothelialization after thermal injury, and increased leukocyte adhesion to the vascular endothelial layer [119, 129]. In contrast, apoER2-deficient mice were protected from these effects by aPL. A requirement for apoER2 in aPL-induced thrombus formation has also been determined in mouse models of APS by two laboratories independently [119, 126]. Our group has shown that aPL administration enhances thrombus formation in the mesenteric arterioles in wild-type mice, whereas it does not do so in apoER2 knockout mice [119]. Using eNOS knockout mice, we further demonstrated that eNOS antagonism is likely an underlying cause of aPL-induced thrombosis. The other group has also demonstrated that apoER2-deficient mice are protected from aPL- or dimerized β2GPI-induced thrombosis in femoral vein [126]. The latter study also used soluble form of the apoER2 binding domain 1 (sBD1), a peptide mimicking the first ligand binding domain which blocks interactions with β2GPI, to further verify that aPL-β2GPI interaction with apoER2 mediates aPL actions in vivo. In addition to propensity for developing thrombosis, APS patients have elevated risk for non-thrombotic vascular occlusion [130–133] and greater stenosis of the celiac, intracranial, mesenteric, and renal arteries compared to non-APS individuals [134–140]. We have recently reported that when medial hypertrophy and neointima formation are invoked in mice by carotid artery endothelial denudation, aPL administration induces exaggerated neointima formation compared to control human IgG [129]. We further demonstrated that the neointima hyperplasia induced by aPL is related to impaired reendothelialization after injury. In contrast to the wild-type animals, apoER2-deficient mice were protected from the adverse effects of aPL on reendothelialization. Inhibitory effect of aPL on endothelial repair was prevented by concurrent administration of molsidomine, exogenous NO donor. These results indicate that aPL exacerbate both thrombosis and non-thrombotic neointima hyperplasia in the mouse models of APS and that the effect of aPL is caused by endothelial dysfunction mediated by aPL-β2GPI interaction with apoER2 (Fig. 6.3A).
In the realm of pregnancy complications in the APS, numerous studies in trophoblast-derived cell lines, human primary trophoblasts, or placental explants have established that aPL treatment attenuates cell proliferation and migration, increases apoptosis, and impairs differentiation by altering the expression or activation of key proteins such as matrix metalloproteinases, interleukins, and chorionic gonadotropin [141]. Abundant expression of apoER2 mRNA was found in the placenta, and our recent study has indicated that both human and mouse trophoblasts express apoER2 protein [80, 107]. The study has further determined for the first time that apoER2 is required for aPL-induced trophoblast dysfunction in culture and fetal loss and IUGR in mice [80]. In cultured trophoblasts, the receptor was required for aPL inhibition of epidermal growth factor (EGF)-induced Akt phosphorylation, cell migration, and proliferation. In mice, pregnant apoER2-deficient mice injected with human aPL showed attenuated fetal loss and IUGR compared to the wild-type mice. In addition to impaired trophoblast function, endothelial cell dysfunction in the placenta also influences pregnancy outcome through insufficient development of endometrial angiogenesis [142, 143]. It has been reported in cultured endometrial endothelial cells and in a mouse angiogenesis model that aPL impair endothelial cell migration and neovascularization [144]. The inhibitory action of aPL was associated with decreased expression of VEGF and matrix metalloproteinases, and it was abrogated by the synthetic peptide TIFI, a competitive blocker of aPL binding to endothelium [144, 145] (Fig. 6.1B). Our work showed in aortic or carotid artery endothelium that aPL inhibit endothelial cell migration, which is dependent on the presence of apoER2 [129]. The requirement for apoER2 in aPL-induced pregnancy loss has been shown in the mice with global apoER2 deficiency, and further studies are warranted to determine whether apoER2 in trophoblast, endothelium, or platelets (or in all three cell types) is specifically required for aPL-induced pregnancy morbidity. Interestingly, in humans polymorphisms in apoER2 gene have been associated with fetal growth restriction [146], although the role of the polymorphism in the pregnancy phenotypes in APS has not been explored.
In summary, aPL binding to β2GPI activates apoER2 to disturb normal cellular functions in endothelial cells and trophoblasts, contributing to enhanced thrombosis and non-thrombotic vascular occlusion as well as adverse fetal outcome in mice. We have recently developed a monoclonal antibody, which inhibits aPL-induced formation of β2GPI-apoER2 complex [147] (Fig. 6.1A). We have shown that the monoclonal antibody decreases thrombus formation and fetal loss in the mouse model of APS. Thus, apoER2 and its downstream effector molecules may provide a new mode of therapeutic interventions to combat APS.
Do Obstetric and Thrombotic Antiphospholipid Antibodies Have Differential Effects upon Target Cells?
What Lessons Have Been Learned from Observational Clinical Studies ?
Current criteria tests used to identify persistently positive aPL in patients with APS do not predict specific APS manifestations [148]. Some patients with these aPL will develop only thrombosis; others only pregnancy morbidity, while some may not develop APS at all [149]. It is not clear why this discrepancy occurs, and one way in which to interpret these findings is that aPL from patients with obstetric APS may bind different antigens and cellular receptors to affect tissues of the body in different ways to aPL from patients with thrombotic APS.
Historically, pregnancy complications in patients with APS were considered to be due to thrombotic events at the maternal-fetal interface. Histological comparison, however, of products of conception from aPL-positive and aPL-negative patients with recurrent early miscarriage has shown a specific defect in decidual endovascular trophoblast invasion in patients with APS [150] and that placental infarction is not specific to patients with APS [151]. Increasing evidence (reviewed above) shows aPL to have inflammatory effects on endometrial and trophoblast cells resulting in impaired implantation and placental development.
Cohort studies show that some patients with APS experience only thrombosis or only pregnancy morbidity but not both. In a European study of 1000 unselected patients with definite APS [152], there were 820 female patients all of reproductive age. Of these patients, 590 experienced obstetric APS [153] and 230 (28% of the entire female cohort) experienced thrombotic APS alone. During a 10-year follow-up period, the most frequent manifestation was thrombosis with thrombotic events appearing in 166 (16.6%) patients during the first 5-year period and in 118 (15.3%) patients during the second period. In the same 10-year period, 127 (15.5%) women became pregnant with a total of 188 pregnancies and 137 live births [154]. The most frequent obstetric complication was early (<10 weeks) pregnancy loss in 16.5% of pregnancies. Interestingly, only three out of 121 (2.5%) women with pure obstetric APS manifestations at study onset developed a new thrombotic event during the first 5-year study period [149].
To enable a detailed analysis of long-term clinical and laboratory characteristics of patients with pure obstetric APS, a European registry has been set up and reported 5-year follow-up data from 247 patients fulfilling only obstetric APS classification criteria at recruitment [155]. Despite further obstetric complications reported in 129/247 (52.2%) patients, there were very few venous – gestational (1.21%) and puerperal (6.7%) – thrombotic events and only three postpartum arterial thrombotic events, two coinciding with abrupt cessation of heparin and aspirin.
Are There Distinguishing Binding Properties of Antiphospholipid Antibodies in Obstetric Antiphospholipid Syndrome?
Many studies have sought to determine whether specific aPL binding properties are associated with thrombotic or obstetric manifestations. The largest study of adverse pregnancy outcomes in SLE is the PROMISSE study. This prospective multicenter study has assessed the frequency of adverse pregnancy outcomes and clinical and laboratory variables that predict them, in women with aPL and/or SLE at conception. To date this study has reported that LA, rather than aCL or aβ2GPI, positivity is the primary predictor of adverse pregnancy outcomes in two independent groups (n = 144 and n = 44) of aPL-positive patients [156, 157].
The association, however, of specific aPL with particular clinical manifestations in established APS cohorts is less certain. The Euro-APS and Euro-obstetric APS studies did not find that specific criteria aPL distinguished thrombotic or obstetric APS. In fact, the Euro-obstetric APS cohort found identification of all three criteria aPL individually or in different combinations to be important in identifying obstetric APS [155]. Overall, no one criteria aPL has emerged as the leading predictor of thrombotic or obstetric manifestations in patients with APS.
It is not clear whether antibodies directed against non-criteria aPL may be more specific for pregnancy loss APS. A recent systematic review was unable to examine whether any non-criteria aPL evaluated (including anti-DI and various IgA aPL) were more frequent in particular APS features because none of the selected studies gave a breakdown of this information and only reported overall prevalence figures for APS [158]. A recent multicenter cohort study [159] that measured IgG, IgM and IgA aCL, aβ2GPI, and anti-DI in APS and controls found IgG aPL to be the commonest and highest titer aPL, while IgA aβ2GPI and anti-DI correlated more strongly with APS compared to IgM counterparts. IgG aCL, aβ2GPI, anti-DI, and IgA anti-DI were associated with thrombotic, but not obstetric complications in patients with APS. Therefore, a convincing association between non-criteria aPL and specific APS manifestations also remains to be proven.
Are There Differences in the Cellular Effects of Obstetric and Thrombotic Antiphospholipid Antibodies?
Experimental evidence presented throughout this chapter highlights how pathogenic aPL interact with β2GPI and/or different cell surface receptors to activate inflammatory pathways leading to the manifestation of obstetric APS. Few studies, however, have specifically set out to compare effects of aPL from patients with and without thrombosis on target cells. Lopez-Pedrera et al. compared monocytes extracted from blood of (n = 44–62) patients with APS with healthy control (HC) monocytes and found differences in p38MAPK and NFκB signaling pathways as well as tissue factor, VEGF, sFlt-1, and PAR1 and PAR2 expression [160–162] in monocytes from APS patients with thrombosis than in monocytes from APS patients with no thrombosis. Lambrianides et al. [163] found that IgG from these two clinical groups in (n = 27) patients with APS had different effects on p38MAPK and NFκB activation in monocytes.
Similarly, few studies have compared the effects of thrombotic versus non-thrombotic APS-IgG in cell types relevant to obstetric APS. Mulla et al. [70] demonstrated that IgG purified from patients with obstetric APS stimulated trophoblast production of IL-8 and GRO-α significantly more than IgG from patients with thrombotic APS [70]. Immunoglobulin G purified from patients with obstetric APS triggered significantly more sEndoglin secretion than IgG from patients with thrombotic APS, while thrombotic aPL induced a greater trophoblast VEGF and sFlt-1 response in the than obstetric aPL [164]. Another comparative study found that only IgG from patients with pure obstetric APS inhibit in vitro trophoblast invasion in a TLR4-dependent manner, compared with thrombotic APS-IgG which lacked this effect [81].
Other studies have utilized proteomics to identify novel pathways in different manifestations of APS. Lopez-Pedrera et al. [165] using traditional proteomics techniques identified 22 proteins that were altered significantly in monocytes of patients with APS (32 with thrombosis and 19 with pregnancy morbidity alone) compared to controls. They found proteins implicated in recurrent spontaneous abortion to be significantly dysregulated in patients with obstetric APS, while six proteins that were most significantly altered among monocytes from patients with thrombotic APS were all functionally related to the induction of a procoagulant state. Ripoll-Nunez et al. [166] utilized newer proteomics techniques to analyze human monocytes treated with IgG from (n = 27) patients with different manifestations of the APS. They found that four of the most significantly regulated proteins were differentially regulated in monocytes treated with thrombotic or obstetric APS-IgG, compared with HC-IgG, thus providing further evidence that the monocyte proteome is differentially regulated by obstetric compared with thrombotic APS-IgG.
As discussed, engagement of the apolipoprotein E receptor 2 (apoER2) has been shown to be required for the adverse effect of aPL on pregnancy outcomes in mice [80]. This effect, however, is not specific to obstetric APS since ApoER2 has also been implicated in aPL-mediated endothelial cell activation [119, 129] and aPL-mediated thrombosis in mice [126].
In summary, evidence is emerging from a variety of clinical and laboratory studies to support the hypothesis that obstetric APS may be distinguished from thrombotic APS by more than just the initial pattern of clinical manifestations. In particular, long-term follow-up reveals little overlap between obstetric and thrombotic manifestations in patients with obstetric APS. Furthermore, there are demonstrable differences in the functional effects of obstetric compared with thrombotic APS-IgG in vitro upon various relevant target cells. Further in vivo work, however, is required to fully answer the question of whether obstetric, and thrombotic aPL have different effects on target cells in patients with APS.
Targeting β2-Glycoprotein I to Prevent Obstetric Antiphospholipid Syndrome
There is sound evidence that β2GPI is present both at the maternal (decidual and uterine endothelial cells) and fetal side (trophoblast cells) acting as an antigenic target for β2GPI-dependent pathogenic aPL in obstetric APS [6, 167].
Related posts:
 Recent Advances in Understanding of the Genetics of Antiphospholipid Syndrome
Clinical and Prognostic Significance of Non-criteria Antiphospholipid Antibody Tests
Prevention and Treatment of Obstetric Antiphospholipid Syndrome
15th International Congress on Antiphospholipid Antibodies Task Force on Catastrophic Antiphospholipid Syndrome Report
Recent Advances in Understanding of the Genetics of Antiphospholipid Syndrome
Clinical and Prognostic Significance of Non-criteria Antiphospholipid Antibody Tests
Prevention and Treatment of Obstetric Antiphospholipid Syndrome
15th International Congress on Antiphospholipid Antibodies Task Force on Catastrophic Antiphospholipid Syndrome Report
 Mechanisms of Antiphospholipid Antibody-Mediated Thrombosis
Mechanisms of Antiphospholipid Antibody-Mediated Thrombosis
 Treatment of Non-criteria Manifestations in Antiphospholipid Syndrome
Treatment of Non-criteria Manifestations in Antiphospholipid Syndrome
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


