Fig. 13.1
Mechanisms of acquired resistance to EGFR-TKI
13.2.1 The T790M Secondary Mutation
Third-generation EGFR-TKIs such as osimertinib (AZD9291) and CO-1686 have shown promising activity in treatment-resistant EGFR mutation-positive NSCLCs containing the T790M mutation [20, 21]. However, acquisition of an EGFR C797S mutation has been identified as a novel mechanism of resistance to osimertinib (AZD9291) [22].
13.2.2 HGF-MET Activation
An additional 10–20 % of tumors from refractory EGFR-mutant NSCLC patients undergo MET gene amplification, which causes HER3-dependent activation of the signaling cascade downstream of EGFR despite its inhibition by TKIs [15]. Recently, MET inhibitors have been administered to NSCLC patients who are naïve or resistant to EGFR-TKIs [23]. This phase II study showed that PFS was extended in the group treated with erlotinib plus the MET inhibitor tivantinib than in the group treated with erlotinib alone, especially among patients harboring KRAS mutations [23]. Yano et al. also showed that HGF, a ligand of the MET oncoprotein, induces gefitinib resistance in EGFR-mutant lung adenocarcinoma cells by restoring PI3K/AKT signaling via phosphorylation of MET, but not EGFR or ErbB3 [16]. Inhibition of HGF-MET signaling may be a useful strategy to overcome resistance to EGFR-TKIs depending on the status of HGF-MET signaling.
13.2.3 Small Cell Lung Cancer Transformation
Morphological transformation to SCLC from NSCLC represents one of the mechanisms of acquired resistance to EGFR-TKIs. Sequist et al. reported the morphological transformation of five drug-resistant NSCLC tumors (14 % of a total of 37 patients) containing EGFR mutations into SCLC [18]. The existing EGFR mutation was maintained during SCLC transformation in all cases [18]. These transformed SCLC tumors were sensitive to standard SCLC chemotherapy.
13.2.4 Epithelial-Mesenchymal Transition
EMT is a progressive biological phenomenon that includes loss of epithelial cell adhesion and induction of a mesenchymal phenotype. Several studies have demonstrated that EMT is associated with reduced drug sensitivity and acquisition of resistance to EGFR-TKIs in NSCLC, whereas retention of an epithelial phenotype ensured a good response to EGFR-TKIs [24, 25]. Recent studies revealed that overexpression of AXL led to resistance to EGFR-TKI in NSCLC cells undergoing EMT and that AXL was a potential therapeutic target in patients with acquired resistance to EGFR-TKIs [19]. These reports suggested that EMT might be a mechanism of resistance to EGFR-TKI. However, the molecular mechanisms of the development of EMT-related resistance to EGFR-TKI are still not fully understood. Therapeutic strategies aiming to prevent EMT with a view to restoring sensitivity to EGFR-TKIs remain to be investigated.
13.2.5 Cancer Stem Cell Properties
Cancer stem cells (CSCs) are characterized by the capacity for pluripotency and self-renewal and are thought to represent a renewable source of cancer cells. The significance of CSC-like properties to the mechanism of resistance to EGFR-TKIs in NSCLCs has recently been investigated [26, 27]. Sharma et al. identified a drug-tolerant cancer cell subpopulation (DTP) that maintains viability under conditions where the vast majority of the cell population is rapidly killed following treatment with gefitinib [26]. The putative CSC marker CD133 was shown to be overexpressed in these DTPs, suggesting a CSC-like phonotype. Shien et al. established 13 gefitinib-resistant, EGFR-mutant NSCLC cell lines. Four of the latter lines showed an EMT phenotype and CSC-like properties, accompanied by overexpression of the CSC markers ALDHA1, ABCG2, and CD44 [27]. These findings may provide clues to overcoming resistance to EGFR-TKIs.
13.2.6 PTEN Loss, FAS/NF-κB Activation, and CRKL Overexpression
Loss of PTEN expression has been associated with decreased sensitivity to EGFR-TKIs owing to activation of PI3K-AKT signaling, impairment of ligand-induced ubiquitination, and degradation of activated EGFR in EGFR-mutant cells [17].
A recent study showed that knockdown of FAS and NF-κB enhanced cell death induced by the EGFR-TKI erlotinib in EGFR-mutant lung cancer cells [28]. Increased expression of the NF-κB inhibitor IκB predicted an improved response and PFS in EGFR-mutant NSCLC patients who received EGFR-TKI therapy [28]. These findings suggest that simultaneous inhibition of EGFR and NF-κB may be useful for the treatment of EGFR-mutant NSCLC.
CRKL overexpression induced resistance to EGFR-TKI mediated by ERK and AKT signaling in EGFR-mutant NSCLC cells [29]. CRKL amplification was found in a lung adenocarcinoma treated with an EGFR inhibitor [29]. These results suggest that CRKL is a therapeutic target for a subset of EGFR-mutant NSCLCs that harbor CRKL amplifications.
13.2.7 MicroRNA Alterations
MicroRNAs (miRNAs) can function as either tumor suppressors or oncogenes and are used as diagnostic, prognostic, and therapeutic biomarkers in lung cancer. Four miRNAs (miR-30b, miR-30c, miR-221, and miR-222) have been shown to play important roles in gefitinib-induced apoptosis and EMT in NSCLC cells in vitro [30]. Several reports demonstrated that EGFR-activated miR-21 is a potential therapeutic target in tumors with mutations in EGFR [31, 32]. Members of the miR-200 family, targeting the E-cadherin suppressors ZEB1 and ZEB2, have been recognized as key suppressors of EMT associated with the resistance to EGFR-TKIs [27, 32]. These findings suggest that miRNAs may be promising therapeutic targets in EGFR-mutant NSCLC patients.
13.2.8 Intrinsic Resistance
Recent studies have identified molecules associated with intrinsic resistance to EGFR-TKIs. BIM (BCL2L11) is a member of the Bcl-2 family encoding a proapoptotic protein. Upregulation of BIM is required for apoptosis induction by EGFR-TKIs in EGFR-mutant NSCLCs. Notably, a BIM deletion polymorphism occurs naturally in 13 % of East Asian individuals. This polymorphism can mediate intrinsic resistance to and reduced responses to EGFR-TKIs in EGFR-mutant NSCLC patients [33]. PFS in response to first-line EGFR-TKIs was significantly shorter in patients with the BIM deletion polymorphism than in those patients with wild-type BIM (8.6 and 4.6 months, respectively [p = 0.004]) [34]. An HDAC inhibitor could restore BIM function and resistance to EGFR-TKI [35]. Treatment with an HDAC inhibitor combined with an EGFR-TKI could represent an attractive strategy to treat EGFR-mutant NSCLC patients harboring a BIM deletion polymorphism.
Yamaguchi et al. demonstrated that thyroid transcription factor-1 (TTF-1)-induced ROR1 was required to sustain EGFR survival signaling as an “Achilles heel” in lung adenocarcinoma. ROR1 could activate kinase-dependent c-Src as well as kinase-independent EGFR-ErbB3, ErbB3 phosphorylation, and PIK3 signaling [36]. Inhibition of ROR1 expression could also restore the sensitivity to the EGFR-TKI. ROR1 expression was evaluated in erlotinib-pretreated tumor samples from 45 EGFR-mutant patients in the EURTAC trial to assess its potential as a predictive biomarker of PFS and overall survival (OS) [6, 37]. The PFS of patients with elevated ROR1 expression was significantly shorter than in those patients with low/intermediate ROR1 expression (11.8 months and 5.8 months, respectively) [37].
Noro et al. recently reported that MET gene amplification could predict short PFS and overall survival (OS) after gefitinib treatment in lung adenocarcinoma (LADC) harboring EGFR mutations [38]. MET-FISH-positive LADC patients defined by MET amplification and gene copy number gains (CNGs) exhibited significantly shorter PFS and OS than patients who were MET-FISH negative [38].
These findings suggest that BIM, ROR1, and MET gene status are involved in intrinsic resistance to EGFR-TKIs and may thus be predictive biomarkers for selecting patients who would benefit from EGFR-TKI therapy.
13.2.9 Future Therapeutic Strategies
Understanding mechanisms of intrinsic and acquired resistance to EGFR-TKI, followed by the development of drugs targeted to molecules that can overcome this resistance, could serve as an important advance for targeting EGFR which is activated in NSCLC. Third-generation EGFR-TKIs such as osimertinib (AZD9291) and CO-1686 showed promising activity in EGFR mutation-positive NSCLCs harboring the T790M mutation [20, 21]. However, an EGFR C797S mutation arose as a novel mechanism of resistance to osimertinib (AZD9291) [22]. EGFR-TKIs combined with platinum-doublet chemotherapy showed prolonged PFS in EGFR-mutant NSCLCs in several clinical trials [39, 40]. A recent study demonstrated that the combination of erlotinib plus bevacizumab could be an effective first-line regimen to treat EGFR mutation-positive NSCLC patients [41]. PFS was significantly prolonged in patients receiving erlotinib plus bevacizumab compared to those that received erlotinib alone (16.0 months and 9.7 months, respectively). There are various mechanisms of acquired resistance to EGFR-TKIs. Future studies should clarify whether there exist other as yet unidentified mechanisms associated with acquired resistance to EGFR-TKI. In addition, treatments of individual patients should be based on assessment of the details of particular mechanism of resistance observed in the patient using re-biopsy or liquid biopsy samples.
13.3 Mechanisms of Resistance to ALK-TKIs
NSCLC patients harboring an ALK fusion gene have shown a strong response to ALK-TKIs such as crizotinib, alectinib, and ceritinib [8–10]. Based on the clinical data, first-line treatment with crizotinib has become the standard therapy for NSCLC patients with ALK fusion gene. However, acquired resistance to ALK-TKI remains virtually inevitable.
13.3.1 Crizotinib
Two major mechanisms of resistance to crizotinib have been demonstrated [42–47] (Fig. 13.2). ALK signal-dependent activation such as ALK secondary mutations and amplification has been reported in half of resistant tumors. The other mechanism is the activation of alternative survival signaling pathways including those mediated by EGFR, KRAS, cKIT, and/or IGF-1R. However, about 20–30 % of resistance mechanisms have yet to be identified.
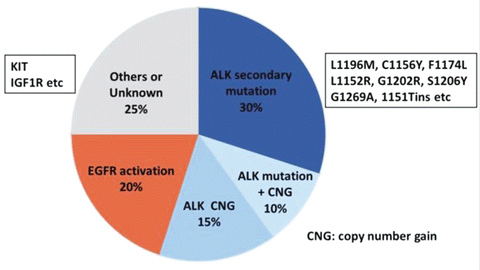
Fig. 13.2
Mechanisms of acquired resistance to crizotinib
13.3.1.1 ALK Secondary Mutations and Amplification
Approximately 30 % of ALK-positive NSCLC patients who have acquired resistance to crizotinib exhibit ALK secondary mutations both inside and outside the gatekeeper site, in analogy with the EGFR-TKI-resistant T790M mutation [42, 43]. Compared with the single T790M gatekeeper mutation of EGFR-TKIs, ALK-positive NSCLC tumors showed multiple gatekeeper mutations (L1196M, C1156Y, F1174L, L1152R, G1202R, S1206Y, G1269A, 1151Tins, etc.) after treatment with crizotinib. ALK gene fusion copy number gain (CNG) was also demonstrated as a mechanism of resistance to crizotinib in vitro [44]. Doebele et al. analyzed re-biopsied samples obtained from 11 ALK gene-rearranged NSCLC patients showing resistance to crizotinib and identified ALK secondary mutations in 36 % of the samples [43]. Two patients (18 %) exhibited ALK CNG [43]. One patient contained both an ALK secondary mutation (G1269A) and CNG [43]. Thus, ALK signal-dependent activation including ALK secondary mutations and amplification is recognized as a mechanism of resistance to crizotinib. Second-generation ALK inhibitors including alectinib and ceritinib exhibit substantial inhibitory potential against tumors with ALK secondary mutations and have been approved in Japan and the Food and Drug Administration (FDA) [9, 10].
13.3.1.2 EGFR Activation
Activation of alternate survival pathways is a major mechanism of resistance to ALK-TKIs. Sasaki et al. demonstrated that EGFR signaling and secretion of EGF and amphiregulin, which are EGFR ligands, are involved in the resistance of H3122 ALK-positive lung cancer cells to crizotinib [45]. EGFR activation could be induced by increased expression of several EGFR ligands, including TGF-α, HB-EGF, and NRG1 as well as EGF and amphiregulin [42, 46]. Almost all mechanisms of EGFR signal-dependent resistance retain ALK signaling. Previous studies reported that crizotinib therapy combined with EGFR-TKIs might be effective against tumors exhibiting EGFR signal-dependent resistance [42, 45].
13.3.1.3 KRAS Mutation, cKIT Amplification, and IGF-1R Activation
Activation of alternate survival signaling pathways such as those mediated by KRAS, cKIT, and/or IGF-1R has also been reported as a mechanism of resistance. KRAS mutations (G12C and G12V) were reported in 2 of 11 ALK-positive NSCLC patients using re-biopsied samples [43]. Two samples derived from crizotinib-resistant NSCLC patients showed amplification of the cKIT gene [42]. Treatment with the cKIT inhibitor imatinib restored the sensitivity to crizotinib. A recent study reported increased expression of IGF-1R and IRS-1 (an adaptor protein that binds to IGF-1R and ALK) in ALK fusion-positive NSCLC patients after crizotinib treatment [47]. Combined treatment with crizotinib and an IGF-1R inhibitor is being considered to overcome resistance to crizotinib in these patients.
13.3.2 Alectinib
Alectinib is a selective ALK inhibitor that was approved in Japan in 2014 [9]. Alectinib has substantial inhibitory potential against tumors with ALK gatekeeper secondary mutations (F1174L, L1196M, L1152R, C1156Y, 1151Tris, and G1269A). However, alectinib showed less efficacy against tumors harboring G1202R mutations. V1180L and I1171T ALK gatekeeper mutations were nevertheless reported as the mechanism of acquired resistance to alectinib [48, 49]. The second-generation ALK inhibitors, ceritinib and AP26113, were effective against ALK-positive NSCLC tumors harboring V1180L and I1171T mutations [48]. HGF/MET signal activation was found in alectinib-resistant NSCLC patients [50]. Crizotinib might be effective against alectinib-resistant NSCLC tumors exhibiting MET activation.
13.3.3 Ceritinib
Ceritinib is a second-generation ALK inhibitor that can block the activity of both ALK and IGF-1R. It was approved by the FDA in 2004 for the treatment of ALK fusion-positive NSCLC patients who failed to respond to crizotinib [10]. Ceritinib could suppress ALK-TKI-induced secondary mutations (L1196M, I1171T, S1206Y, and G1269A) and overcome the resistance to alectinib associated with I1171T and V1180L secondary mutations [51]. However, ALK secondary gatekeeper mutations (L1196M and G1269A) were also observed in ceritinib-resistant NSCLC cells [52, 53]. These resistances could be overcome by alectinib [52, 53].
13.3.4 Future Therapeutic Strategies
The response of ALK-positive NSCLC patients to ALK-TKIs is well documented, although the development of drug resistance is a challenge that must be overcome. Compared with the T790M gatekeeper mutation of EGFR-TKIs, ALK-positive NSCLC tumors developed multiple gatekeeper mutations following treatment with ALK-TKIs. The second-generation ALK-TKIs alectinib and ceritinib could overcome these gatekeeper mutations, whereas these TKIs might paradoxically induce these mutations. In addition, novel second-generation ALK-TKIs including AP26113, ASP3026, and TSR-001 are being developed and are expected to overcome drug resistance. Two randomized phase III trials (the J-ALEX and ALEX studies) are designed to evaluate the efficacy and safety of alectinib compared with crizotinib in ALK-positive NSCLC patients. The question of timing and the sequence of different ALK-TKIs treatments will be answered based on the results of these trials. Furthermore, ALK-TKIs combined with standard chemotherapy or molecular-targeted agents are being considered as a therapeutic strategy to overcome drug resistance. Evidence of chemotherapy with ALK-TKIs in ALK-positive patients has not yet been shown; however, ALK-TKI combination therapy is being planned for the future.
Related posts:
 Small Cell Lung Cancer and Molecular Targeted Therapy
Management of Adverse Effects by Molecular Targeted Therapy and Immunotherapy
Small Cell Lung Cancer and Molecular Targeted Therapy
Management of Adverse Effects by Molecular Targeted Therapy and Immunotherapy
 Screening Lung Cancer with Low-Dose CT Combined with Molecular Markers
Screening Lung Cancer with Low-Dose CT Combined with Molecular Markers
 Health-Related Quality of Life in Molecular Targeted Therapy
Health-Related Quality of Life in Molecular Targeted Therapy
 EGFR Mutant
EGFR Mutant
 PET-CT, Bio-imaging for Predicting Prognosis and Response to Chemotherapy in Patients with Lung Cancer
PET-CT, Bio-imaging for Predicting Prognosis and Response to Chemotherapy in Patients with Lung Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


