Fig. 13.1
Mechanisms for the intergenerational transmission of programming effects. (a) Persistence of an adverse external environment can result in the reproduction of the phenotype in multiple generations. (b) The induction of programmed effects in the F1 offspring following in utero exposure (e.g., programmed changes in maternal physiology or size) results in programmed effects on the developing F2 fetus and so on (Reproduced with permission Drake and Walker [35])
Experimental animal studies have produced similar generational effects when environmental stressors are placed upon successive generations of offspring. In a mouse study that used a persistent high-fat diet for each generation, F0 grandmothers and F1 mothers (all fed a high-fat diet during pregnancy and lactation) produced F2 male offspring that were also shown to be predisposed to obesity and hepatic steatosis [42]. In a study using rats that were overfed after birth, Plagemann and colleagues [43] purport an epigenetic model of obesity and the metabolic syndrome that is underpinned by the developmentally programmed dysregulation of hypothalamic body weight regulation. They conclude, based on their findings, that the “vicious intergenerative circle” of obesity and cardiometabolic disorders remains closed unless the maternal diet is modified during the nursing period ([43], p4974) (Fig. 13.1).
13.4 Maternal Environment Effects
Researchers have found that a single maternal (F0) environmental exposure can produce effects in F1 and F2 generation offspring via developmental programming. These observations are distinct from the developmentally programmed changes found only in the generations of offspring that are directly exposed to the stressed maternal environment (as discussed in the previous section). Thus, Skinner [44] has put forth that persistent exposure to environmental conditions should be referred to as a “multigenerational phenotype,” distinguishable from “transgenerational inheritance” that is transmitted epigenetically via the germline (see below). In the case of the multigenerational phenotype, when a pregnant mother is consuming an inadequate, low-protein diet, three generations are effectively being exposed simultaneously to this dietary insult: the pregnant mother (F0), her fetal offspring (F1), and the primordial germ cells (PGCs)—the precursors of sperm and eggs—within the F1 fetus (Fig. 13.2).
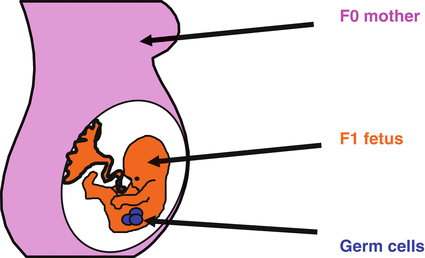
Fig. 13.2
Multigenerational exposure to an environmental effect. An environmental insult during pregnancy to a mother (F0 generation) might affect not only the developing fetus (F1 generation) but also the germ cells which will go on to form the F2 generation (Reproduced with permission Drake and Walker [35])
As discussed previously, it has been well established in experimental animal studies and epidemiological literatures that a connection exists between low birth weight and subsequent adult risk for cardiometabolic syndrome [4, 45]. “Maternal constraint” was one of the first “maternal effects” studied by researchers. The maternal constraint of birth weight has been described in both maternal “supply” and “demand” terms [46]. Maternal constraint in terms of supply may be due to decreased uteroplacental blood flow, reduced blood volume, decreased oxygen-carrying capacity, prenatal nutrition, teratogens, and short birth intervals. Demand factors limiting fetal growth include such things as multiple gestations [47]. By using artificial insemination to cross small Shetland horses and large, draft, Shire horses, Walton and Hammond [48] demonstrated in their renowned crossbreeding study that offspring birth size was largely governed by the size of the gestating mother. In other words, smaller Shetland mothers gave birth to small foals regardless of parental genotype. A 37-year-long epidemiological study in Norway found concurring results. By analyzing birth weight, the researchers discovered that genetic factors accounted for only 31 % of birth weight variation [49]. Furthermore, several other population-based studies compared maternal birth weight and paternal birth weight with offspring birth weight and found the maternal effect to be considerably stronger [49–52], although there have been a few report findings to the contrary [53]. In the Cebu, Philippines, cohort study, however, maternal intergenerational birth weight correlations were not found to be stronger among taller women [50]. The authors surmised that reduced adult stature caused by a history of nutritional insufficiency did not lead to a further increased constraint of offspring birth weight. According to the authors, some of the mechanisms that might explain their findings include sex-linked genetic effects and indirect genetic, epigenetic, and shared environmental/cultural effects. It was noted that in every case the mechanism exerted its effect on birth weight for both the mother and child in utero [50]. In sum, maternal effects that disproportionately affect birth weight are complex and are probably influenced by a number of factors that reach well beyond adult maternal size and stature.
There is also corroborating evidence from human cohort studies concerning multigenerational maternal environment effects. In the Dutch “hunger winter” studies, it was found that mothers who were exposed to the famine during pregnancy (living on as little as 500 kcal/day) had female offspring (F1) with dysregulated lipid profiles (i.e., cholesterol and triglycerides) as opposed to offspring not exposed to famine conditions in utero [54]. They further found that the children of the exposed F1 females exhibited higher rates of neonatal obesity and were nearly twice as likely to develop cardiometabolic disease than the unexposed control group [55].
Multigenerational (i.e., F1, F2) effects have been documented through extensive experimental animal studies as well [36]. Using mostly rodent models, researchers have shown that a single (F0) maternal exposure generates cardiometabolic programming effects on F1 and F2 animals. A number of these studies restricted (F0) mothers to protein and/or energy diets during pregnancy and/or lactation and observed programmed effects in both F1 and F2 generation animals on insulin sensitivity and glucose tolerance [56–58], birth weight, blood pressure, kidney structure [59], and obesity [58]. Other studies exposed F0 mothers to excess glucocorticoids [60] or made surgical alterations to the placenta [61] and observed developmentally programmed effects on birth weight, glucose tolerance, and blood pressure.
Multigenerational effects linked to prenatal exposures to restrictive maternal diets are not the only models used in experiments of this kind. In feast pathway studies (as discussed above), researchers examined the cardiometabolic structure and function effects of maternal nutritional excess and/or obesity on offspring prior to conception, during pregnancy, and/or while nursing (see [8]). Studies have shown that multigenerational effects on obesity and other cardiometabolic traits can be tied to initial (F0) maternal nutritional excess. This environment during pregnancy influences body size and insulin sensitivity in both F1 and F2 offspring [62]. In an experiment utilizing mice, Gniuli et al. [63] report that a prenatal high-fat diet, especially if followed by a maternal high-fat diet while nursing, induces a “type 2 diabetes phenotype” in F1 offspring, which can then be transmitted to their F2 progeny, even if additional dietary insults are not provided. Indeed, the perinatal maternal nutritional environment alone has far-reaching implications. Srinivasan and colleagues [64] fed female rat pups a high-fat diet during the suckling period, resulting in obese adults. Their adult F2 offspring also became obese, even though their F1 mothers were fed a standard chow diet after weaning.
It remains unclear what mechanisms account for the intergenerational transmission of these developmentally programmed traits. Possible mechanisms include altered organ development, cellular signaling dysregulation, and epigenetic modifications. Perhaps, at least in some cases, the multigenerational programming effects of both the F1 and F2 generations are not due to the same, single maternal in utero exposure (e.g., F0 maternal low-protein diet) (Fig. 13.3). Alternatively, it could be that cardiometabolic dysfunction resulting from the early environment exposure of the F1 female gives rise to an altered intrauterine environment during her own pregnancy that is distinct from the one in which she was gestated (F0) but that also leads to the same developmentally programmed trait in her (F2) offspring. Indeed, some animal models of multigenerational transmission of glucose intolerance from F1 to F2 generations point to such a sequence. Several rodent studies have shown that glucose-intolerant, adult F1 animals whose mothers (F0) were fed a low-protein diet during pregnancy are insulin sensitive but have insufficient insulin secretory capacity as adults, which in turn results in glucose intolerance [65]. The glucose-intolerant adult (F2) offspring of these (F1) females, on the other hand (whose F1 mothers consumed a control diet during gestation/lactation), are insulin resistant (rather than insulin sensitive) and hyperinsulinemic (as opposed to insulin deficient) [56]. In this example, both F1 and F2 generation animals are glucose intolerant, apparently due to different developmental disruptions in glucose-insulin metabolism and due to distinct developmental programming pathways in the intrauterine environment. In this scenario, the intrauterine perturbation in one generation causes a metabolic cascade that leads to another (similar or dissimilar) cascade in the subsequent generation and so on (Fig. 13.1b).
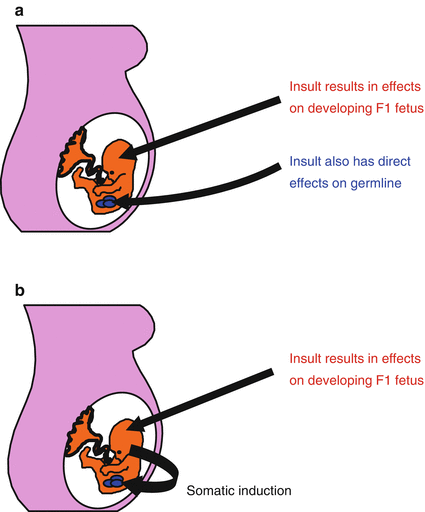
Fig. 13.3
Transmission of epigenetic effects through gametes. (a) An environmental exposure affects the developing F1 fetus but also has direct effects on the developing germ cells which form the F2 generation. (b) Alternatively, effects induced in the developing F1 fetus can be transmitted to the germ cells which will form the F2 generation (somatic induction) (Reproduced with permission Drake and Walker [35])
Importantly, in most of the aforementioned multiple-generation animal studies, the research design did not include an F3 generation. Of the two studies that did include an F3 generation in their research design [59, 60], no evidence of transmission to F3 animals was found. The lack of F3 inheritance might suggest that the observed F1 and F2 effects were due to direct exposure (of the F1 fetus and F2 PGCs) in the (F0) intrauterine environment—lending some support to Skinner’s notion of a “multigenerational phenotype” [44]. Both types of developmental programming inheritance may occur (Figs. 13.1b and 13.2).
13.5 Transgenerational Inheritance: Epigenetic Traits Transmissible Through the Germline
In order for developmentally programmed cardiometabolic traits to be transmitted to the F3 generation and subsequent generations after that (i.e., “transgenerational inheritance”), information about paternal and/or maternal exposure must be transmitted through the germline, since no direct exposure of the initial insult could exist for F3. Epigenetic inheritance research is now uncovering how this non-genomic inheritance may be possible in the germline when there are no changes in genetic structure (i.e., base pairs of DNA).
A relatively rare process of genomic imprinting was discovered in the 1980s [66], putting the epigenetic regulation of gene expression on the map. So although this mechanism has been recognized for some time, the possibility and importance of the transgenerational inheritance of epigenetic marks, and how it works, is only now beginning to be widely accepted and understood. Why did it take decades to come to light? The answer is that the traditional view in developmental biology and genetics held that epigenetic marks are erased and “reset” in the primordial germ cells (PGCs) in each generation. Since the epigenetic germline is reset, it allows the genetic regulatory system to be reestablished in each generation. This commonly known process has put the frequency and importance of epigenetic inheritance into doubt. However, new research discoveries have begun to alter that viewpoint [67, 68]. Through a process of chemical conversion and subsequent dilution during cell division, Hackett and colleagues [69] recently discovered how methylation marks are erased in mouse PGCs. They also determined how some (rare) methylation marks escape erasure, thereby providing a potential pathway for true transgenerational epigenetic inheritance. Researchers, in another breakthrough study, demonstrated that approximately 1 % of histone marks (located at key sequences with gene regulatory functions) remain in mouse sperm after cell differentiation, thereby furthering our nascent understanding of the inner workings of epigenetic inheritance [70].
These new findings may help us understand several epidemiological research discoveries that appear to reflect such processes at work in humans. In one such study, researchers analyzed three Swedish birth cohorts born in the late 1800s and early 1900s who experienced fluctuating food availability. The study’s goal was to determine if food supply during the slow-growth phase (ages 8–2) of paternal grandfathers and grandmothers was linked to the risk of diabetes, cardiovascular disease, and longevity in their children and grandchildren. They discovered that low food intake by fathers during their slow-growth phase was associated with lower risk of diabetes and cardiovascular disease of sons. Conversely, an increased food intake during the paternal grandfather’s slow-growth period was linked to increased risk for diabetes among grandsons [71, 72]. Food supply during the slow-growth phase of paternal grandfathers was associated with lifespan in grandsons, and likewise, food supply during fetal/infant life or the slow-growth period of paternal grandmothers was associated with lifespan in granddaughters [73].
The goal of Soubry and coworkers [74] was to determine if there is a relationship between parental obesity and DNA methylation at the insulin-like growth factor II (IGF-II) gene in their children (IFG-II is linked to a risk for obesity, gestational diabetes, and some forms of cancer). The researchers compared information about parental obesity to methylation patterns found in DNA from the umbilical cords of the fathers’ newborn children. The results showed that the offspring of obese fathers had significantly lower DNA methylation at the IGF-II gene than did the children of nonobese fathers. The data augmented evidence for the transmission of epigenetic effects associated with obesity from males to their offspring.
Some animal studies have shown similar transgenerational effects. Throughout India, Southeast Asia, and Polynesia, chewing betel nuts is a popular practice. The betel nut contains the toxin nitrosamine and is known to increase risk for the cardiometabolic syndrome [75]. Three generations of experimental animals were used to model the effects of betel nut consumption on blood glucose. Boucher and colleagues [76] fed young mice betel nut in their standard chow for 4–6 days and then administered glucose tolerance tests to the F0 animals in adulthood. Consuming betel nut was found to significantly increase the risk of glucose intolerance in F0 animals. They also saw an increased risk in the F1 and F2 offspring of males that consumed betel nuts in their chow before mating.
Three consecutive generations were used in another rodent study to determine the effects of a (F0) maternal high-fat diet on offspring body size and glucose-insulin metabolism. Results revealed that, compared to control animals, both F1 and F2 generation animals were insulin resistant and had increased body length even though they were fed control diets. These effects were seen in both the paternal and maternal lineages. Animals were no longer insulin resistant in the F3 generation, but females inherited increased body length through the paternal line [77].
We [37] also experimented with three generations of rats. F0 female rats were fed a low-protein diet during pregnancy and lactation. The results showed reduced insulin secretory capacity in F1 rats and insulin resistance in their F2 offspring through the maternal line. F3 generation males also exhibited insulin resistance compared to controls, although it was less severe than in F2 animals. Changes in glucose-insulin metabolism in the F1, F2, and F3 generations can be traced to dietary alterations in F0 females, just as in the previous study. After weaning, all animals were fed control diets, as were pregnant and lactating F1 and F2 females. This means that at least some epigenetic effects connected with insulin-glucose metabolism survived germline epigenetic mark erasure by the F3 generation.
While evidence of inherited epigenetic alterations in the F2 generation has been confirmed in other animal studies, they found no evidence of transgenerational effects in F3 animals. Some of these experiments subjected pregnant F0 females to glucocorticoid overexposure [60] and low-protein diets during gestation [59].
The presence of F3 transgenerational epigenetic effects in some studies but not others requires explanation. A number of investigations modeled Skinner’s “multigenerational phenotype” [44] that is due to direct exposure (Fig. 13.2) and therefore may lack F3 effects. In contrast, other studies were set up to allow the capture of “transgenerational inheritance” through the germline, via either direct effects on the developing germ cells in utero (Fig. 13.3a) or indirectly via “somatic induction” of the developing fetus [78] (Fig. 13.3b). Another possibility is that an epigenetic threshold was reached in some studies, but in others it was not. A threshold effect may calibrate to the type, intensity, duration, and/or timing of the initial F0 exposure and may determine the durability of epigenetic marks undergoing intergenerational epigenetic reprogramming. Or perhaps under some conditions, epigenetic marks that initially accumulate in the F1 generation and manage to avoid epigenetic erasure in F2 have become so “diluted” in subsequent generations that the phenotypic traits can no longer be identified. This might explain why some transgenerational studies found programmed effects diminished from F2 to F3 (e.g., 37). Since the reason(s) are still unclear, a comprehensive explanation of the mechanistic differences between multigenerational phenotypes and epigenetic transgenerational inheritance will have to wait for future studies of more than three generations, of which there are very few to date (Fig. 13.3).
13.6 The Intergenerational Transmission of Developmentally Programmed Cardiometabolic Traits: Implications for Prenatal Interventions
The existing worldwide obesity epidemic [79], in tandem with persistent undernutrition and poor food availability [80], emphasizes the importance of research involving the developmental origins and the intergenerational transmission of cardiometabolic dysregulation. The public health challenges associated with the scale of these disorders are staggering, and cost-effective and efficacious interventions are desperately needed. Is it possible that the principles of the developmental origins of cardiometabolic disease can also be leveraged to slow or reverse the current epidemiological trends? Based on the prevailing state of research, there is reason for both optimism and caution. Theoretically, the same physiological bases of developmental plasticity that lead to cardiometabolic dysregulation should be able to prevent, slow, or reverse the current trends. At the same time, developmental plasticity is itself shaped by evolutionary principles and processes that must be understood—and accounted for—if such interventions are to reach their full potential.
It has been over 20 years since the “thrifty phenotype” was first proposed, and today many researchers and public health officials continue to believe that the same developmentally plastic features of cardiometabolism that cause adult disease can be “reverse engineered” to improve health instead. Much of the research has targeted optimized fetal growth, with the goal of improved health later in life. To be sure, the possibilities surrounding the promise of such approaches during pregnancy and early postnatal are well represented in the DOHaD literature [40, 81, 82]. And indeed, a steadily increasing number of clinical trials and pharmacological and, especially, experimental animal studies continue to suggest the potential of interventions targeting the prenatal period to significantly improve cardiometabolic health outcomes later in life [83]. Some of the most promising metabolic syndrome-preventative strategies targeting early growth and development include maternal prenatal dietary supplementation with genistein, (−)-epigallocatechin-3-gallate (EGCG), vitamin D, and omega-3 fatty acids (see Table 13.1). Of these, maternal prenatal supplementation with omega-3 long-chain polyunsaturated fatty acids has been the most thoroughly studied, via experimental animal studies, human observational studies, and clinical trials.
Table 13.1
Metabolic syndrome prevention via prenatal maternal diet
Dietary factor | Findings | References |
|---|---|---|
Genistein | Experimental animal studies suggest that early exposure to genistein may influence the adipogenesis processes and obesity-related diseases such as type 2 diabetes | [84] |
EGCG | Experimental animal studies show maternal prenatal diet supplemented with green tea extract a rich source of (−)-epigallocatechin-3-gallate (EGCG) attenuates high-fat diet-induced insulin resistance in adult male offspring | [85] |
Vitamin D | Several clinical trials have demonstrated improved insulin sensitivity and glucose tolerance among women with GDM when diets are supplemented with vitamin D and calcium | [86] |
Omega-3 fatty acids | Experimental animal studies show improved insulin sensitivity among offspring whose mothers were fed omega-3 rich diets during pregnancy and pregnancy/lactation but were then fed either control or high-saturated-fat diets post-weaning; clinical trials suggest maternal prenatal omega-3 supplementation protects against low-birth-weight phenotype and is associated with improved body composition and insulin sensitivity in neonates |
Several experimental animal studies have investigated the effects of maternal prenatal/lactation diets rich in omega-3 LCPUFAs on systolic blood pressure, as well as levels of serum leptin and insulin in neonatal offspring [91]. Researchers have also investigated adult offspring weaned onto control [87] and “Western” diets high in saturated fat [88]. These studies all found that maternal diets rich in omega-3 LCPUFAs lead to improved cardiometabolic function (e.g., insulin resistance) in adult offspring consuming adequate or more than adequate diets. Observational and prospective studies have also associated higher intakes of omega-3 LCPUFAs during pregnancy with increased gestation duration and birth size (see [92] references) and reduction in deep placentation disorders. These associations, thereby, provide a possible mechanism for improved fetal growth and its associated improved cardiometabolic health outcomes [93]. Zhao and colleagues [94] recently assessed maternal and cord plasma fatty acids in relation to indicators of fetal insulin sensitivity in 108 mother-newborn pairs. Their findings showed that lower circulating levels of docosahexaenoic acid (DHA)—an important omega-3 LCPUFA—were associated with lower fetal insulin sensitivity. Several recent randomized clinical trials have confirmed and extended these findings by demonstrating that maternal prenatal omega-3 supplementation protects against the metabolic syndrome-inducing low-birth-weight phenotype [92] and is associated with improved body composition and insulin sensitivity in neonates [90]. It should be noted, however, that a study of the 19-year-old offspring of mothers supplemented with LCPUFA-rich fish oil during the third trimester of pregnancy failed to show enduring improvements in plasma lipids, blood pressure, or heart rate [95, 96].
13.7 Accounting for Evolution in Future Metabolic Syndrome Prevention Strategies
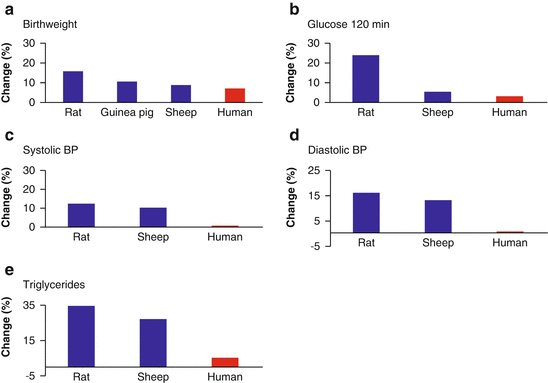
While the studies referred to above do offer hope that the current metabolic syndrome global pandemic might be slowed or reversed, to date, evidence from dozens of observational studies and controlled trials has tempered some of the more unbridled early optimism regarding the potential effectiveness of such interventions. In efforts to improve fetal growth as well as maternal and child health outcomes, researchers in many early studies supplemented maternal diet during pregnancy with targeted macro- and micronutrients. These studies have reported mixed results, however, and even when supplementation interventions were successful, often only small improvements were observed (see [97–99]). Although some of these approaches do appear to have potential [100], it is the species-specific timescales on which the developmental pathways are calibrated that may be to blame for their less than remarkable successes. As Kuzawa and Thayer point out, the “principles of evolutionary biology and adaptation lead us to hypothesize that many short term interventions that trigger large biological changes in short lived species, will have comparably small effects in humans” ([101], p229). In other words, differences in body size, lifespan, variability in the physical environment, and reproductive strategy of various mammalian species (e.g., rodents, guinea pigs, sheep, and humans) exert significant influence on the magnitude of developmentally programmed effects due to early life environments. Because of this, phenotypic plasticity operates under very different constraints for small-bodied, short-lived species that are subject to relatively little physical environmental variation over multiple generations (e.g., rodents) versus large-bodied, long-lived species that often occupy highly variable physical environmental conditions—often over just one reproductive life history stage (e.g., humans). Data from experimental animal studies, for example, assessing the effects—and effect sizes—of perturbations during gestation (e.g., maternal dietary restrictions during pregnancy), provide clear evidence of the consequence of these species-specific evolutionary processes (Fig. 13.4).
 Nutrition Around the Clock and Inflammation: The Right Food at the Right Time Makes the Difference
Nutrition Around the Clock and Inflammation: The Right Food at the Right Time Makes the Difference
 Obesity and Inflammation in Pregnancy
Obesity and Inflammation in Pregnancy
 PCOS and Pregnancy: Impact of Endocrine and Metabolic Factors
PCOS and Pregnancy: Impact of Endocrine and Metabolic Factors
 The Role of Placental Inflammasomes in Linking the Adverse Effects of Maternal Obesity on Fetal Development
The Role of Placental Inflammasomes in Linking the Adverse Effects of Maternal Obesity on Fetal Development
 Gastrointestinal Symptoms and Nutritional Profile During Pregnancy
Gastrointestinal Symptoms and Nutritional Profile During Pregnancy
 Pathology of the Placenta: A Continuum Spectrum of Inflammation from Physiology to Disease
Pathology of the Placenta: A Continuum Spectrum of Inflammation from Physiology to Disease




Related posts:
Nutrition Around the Clock and Inflammation: The Right Food at the Right Time Makes the Difference
Obesity and Inflammation in Pregnancy
PCOS and Pregnancy: Impact of Endocrine and Metabolic Factors
The Role of Placental Inflammasomes in Linking the Adverse Effects of Maternal Obesity on Fetal Development
Gastrointestinal Symptoms and Nutritional Profile During Pregnancy
Pathology of the Placenta: A Continuum Spectrum of Inflammation from Physiology to Disease
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
