Fig. 2.1
Clinical orientation for DSD
In some cases, the external genitalia are “ambiguous” and are associated with:
A genital bud that is midway between a penis and a clitoris
Genital ridges that are incompletely fused (forked scrotum)
The urethral opening on the underside of the penis (hypospadias) or a single perineal orifice at the base of the genital bud, between the genital ridges
Gonads that are impalpable or palpable in the inguinal position
In other cases, the malformation is less clear-cut and attention should be focused on:
Bilateral cryptorchidism
Posterior hypospadias
Cryptorchidism associated with hypospadias
Micropenis (<2.5 cm)
Clitoral hypertrophy
A nonvisible vaginal orifice with posterior fusion of the genital ridges in a newborn with female phenotype
Palpation of a uni- or bilateral mass in the inguinal position or in the labia majora
The degree of the DSD should be evaluated according to the Prader classification (Stage I to V) (Table 2.1).
Table 2.1
The Prader scores
Stage 0
Normal female external genitalia
Stage 1
Female external genitalia with clitoromegaly
Stage 2
Clitoromegaly with partial labial fusion forming a funnel-shaped urogenital sinus
Stage 3
Increased phallic enlargement. Complete labioscrotal fusion forming a urogenital sinus with a single opening
Stage 4
Complete scrotal fusion with urogenital opening at the base or on the shaft of the phallus
Stage 5
Normal male external genitalia
2.1.2 Genetic Investigations
The presence of the SRY gene should be determined with PCR and the results should be ready within 24 h. This will determine whether the case is an undervirilization of a 46,XY fetus or the excessive virilization of a 46,XX fetus. It is essential to determine the karyotype, as this will enable the diagnosis of chromosomal abnormalities like 45X/46,XY mosaicism. However, this type of diagnosis will take from a couple of days to several weeks.
2.1.3 Hormonal Investigations
The hormone work-up should be performed between 6 and 36 h after birth. The following measurements are crucial: 17-hydroxyprogesterone (17-OHP), testosterone (T), and anti-Mullerian hormone (AMH). If possible, follicle-stimulating hormone (FSH), luteinizing hormone (LH), and delta4androstenedione (Δ4A) measurements should be associated.
High 17-OHP suggests congenital adrenal hyperplasia (CAH), which is usually due to a deficiency in 21-hydroxylase that results in a 46,XY DSD.
The values of T and AMH are crucial for evaluating DSD with the 46,XY karyotype. The T, FSH, and LH levels should be reevaluated during the minipuberty (days 15–90), which is the period of activation of the hypothalamic-pituitary-gonadal axis. During other periods, T and its precursors should be evaluated after the human chorionic gonadotropin (hCG) stimulation test. Most often the long test is used (1500 UI 1d/2 × 7). All these investigations should be able to differentiate 46,XY DSD secondary to insufficient androgen production (gonadal dysgenesis or defective androgen synthesis) from those secondary to androgen resistance associated with normal T production.
2.1.4 Imaging
Fetal ultrasonography sometimes reveals a uterus. Genitography can show evidence of Mullerian derivatives. Urogenital endoscopy under general anesthesia can be used to specify the implantation height of the Mullerian cavity when it is present.
2.2 Causes of DSD
According to the 2006 consensus statement of the european society for paediatric endocrinology (ESPE) [1] (Table 2.2), we differentiate between 46,XX DSD, 46,XY DSD, DSD with ovotestis, and DSD associated with chromosome abnormalities.
Table 2.2
Comparison between the old and new DSD classification (ESPE Consensus 2006)
Previous classification | New classification |
|---|---|
Sexual ambiguities | Disorders of Sexual Development (DSD) |
MPH (Male Pseudohermaphrodisms) | 46,XY DSD |
FPH (Female Pseudohermaphrodisms) | 46,XX DSD |
True Hermaphroditism | Ovotestis, DSD |
46,XX male | 46,XX testicular DSD |
45,X0, 46,XX Klinefelter Syndrome 47,XXY | DSD with chromosomic abnormalities |
2.2.1 46,XX DSD
46,XX DSD is due to the masculinization of the 46,XX fetus because of excessive exposure to androgens during intrauterine life, whether of endogenous or exogenous origin. It may also be due to abnormal ovarian determination.
2.2.1.1 Fetal Hyperandrogenism
In the great majority of cases (75 %), the 46,XX fetus has been overexposed to fetal androgens in the context of congenital adrenal hyperplasia (CAH). The excessive adrenal androgens produced upstream of an enzymatic block are peripherally converted to T and dihydrotestosterone (DHT) and cause fetal virilization (Fig. 2.2).
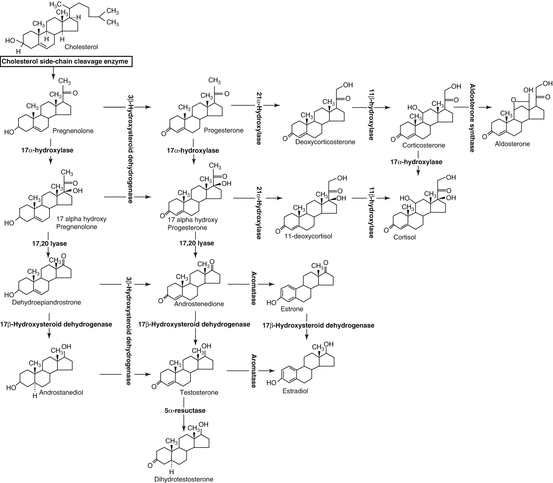
Fig. 2.2
Steroidogenesis
DHT deficiency is the most frequent (90–95 %) of the enzymatic blocks, accounting for 1/14,000 births. Systematic screening with blotting paper is performed on the third day of life. A salt-wasting condition with hyponatremia may be associated with the virilization because of aldosterone and glucocorticoid defects downstream of the enzymatic block. The elevated plasma 17-OHP is generally >50 ng/ml. Treatment with glucocorticoids and mineralocorticoids should be undertaken as soon as possible. Molecular study of the CYP21A2 gene will confirm the diagnosis, since this disorder is recessively transmitted [2]. The sex of rearing of these newborns is almost always female; however, given the excessive androgen exposure during fetal life and the evidence of disturbed gender identification in adulthood, some authors have questioned systematic female orientation in the case of highly virilized CAH [3].
Other forms of CAH can be encountered, though these are rarer. The 11-beta-hydroxylase block is seen in 5 % of the cases. No salt-wasting occurs. The diagnosis is based on simultaneously elevated S-component and desoxycorticosterone (DOC), and a ratio of Δ4/17-OHP >1 is highly suggestive. Study of the CYP11B1 gene will confirm the diagnosis since transmission is autosomal recessive [4]. Careful substitutive therapy with glucocorticoids should be undertaken. The 3-beta-hydroxysteroid dehydrogenase block is rare (1 %). It associates salt-wasting and moderate virilization. During hormonal investigations, high 17-OH pregnenolone is evident and moderately elevated 17-OHP is also frequently noted, probably due to the effect of the hepatic 3ß HSD. An HSD3B2 gene abnormality will confirm the diagnosis, since transmission is recessive [5]. Substitutive therapy with gluco- and mineralocorticoids should be prescribed.
More rarely, the virilization of the 46,XX fetus may be caused by a lipoid adrenal hyperplasia secondary to a mutation of the StAR gene, which codes for the steroidogenic acute regulatory protein (StAR). Transmission is autosomal recessive. The StAR protein is involved in the cholesterol transport in mitochondria, which is the first step in adrenal and gonadal steroidogenesis. Lipoid adrenal hyperplasia is thus characterized by a major deficit in adrenal and gonadal steroidogenesis, evidenced by severe adrenal insufficiency and a female phenotype in both 46,XX and 46,XY fetuses. However, less severe forms – characterized by adrenal insufficiency but less pronounced undervirilization – have recently been reported [6].
The P450-oxidoreductase (POR) deficit is a possible etiology of 46,XY DSD, as well as 46,XX DSD [7]. These forms are dealt with more extensively in the chapter on 46,XY DSD.
2.2.1.2 Exogenous Hyperandrogenism
This type of fetal hyperandrogenism may be secondary to placental aromatase gene mutation, which is recessively transmitted. This abnormality is rare and is characterized by maternal virilization in the third trimester of pregnancy with spontaneous regression after delivery [8]. Another cause of exogenous androgens is one of the rare ovarian tumors, such as luteoma of pregnancy or maternal adrenal tumors [9].
2.2.1.3 Abnormalities in Gonadal Determination
Abnormalities in gonadal determination lead to 46,XX testicular DSD, and affected individuals were formerly termed XX males. These patients may present genital abnormalities during the neonatal period or a normal male phenotype. In the latter case, the diagnosis is made in adult life, frequently because of infertility. Since about 10 % of patients are SRY negative, other genes are probably involved in testis determination. It has been hypothesized that these abnormalities are secondary to either an underexpression of ovary-determining genes or an overexpression of testis-determining genes [10], and both hypotheses have been reinforced by evidence [11]. Moreover, Camerino et al. reported an RSPO1 gene mutation in the family of a 46,XX patient with male phenotype associated with palmoplantar hyperkeratosis [11].
2.2.2 46,XY DSD
46,XY DSD refers to the case of 46,XY newborns with undermasculinization. One or both gonads are usually palpated at birth. The hormonal levels of T, AMH, FSH, and LH, as well as the presence of Mullerian derivatives at pelvic ultrasonography, will differentiate gonadal dysgenesis (associated with insufficient gonadal secretion of T and AMH) from T production defects or T insensitivity.
2.2.2.1 Gonadal Dysgenesis
Gonadal dysgenesis is a defect in testis determination characterized by a variable alteration in Leydig and Sertoli cell function. This disorder may be secondary to mutations in any of the several genes taking part in the differentiation process of the primitive gonad to a testis.
SRY Gene Abnormalities
SRY gene abnormalities express with a clinical picture of 46,XY sex reversal with female phenotype. If gonads are not palpated at birth, it is probable that the diagnosis will be made in the pubertal period in the context of primitive amenorrhea associated with pubertal delay. However, some patients may present partial pubertal development, often caused by an association with a secreting gonadoblastoma [14]. This picture of 46,XY sex reversal is associated with a SRY gene mutation in 20 % of the cases.
Abnormalities in Other Sex Determination Genes
About 80 % of the cases of gonadal dysgenesis are not caused by a SRY gene abnormality. They may be secondary to abnormalities in the other genes that take part in testis determination, however, and they are autosomal or X-linked.
Some cases of gonadal dysgenesis have been linked to SF1 gene mutation. This gene is involved in the development of male gonads and the adrenal glands [15]. The phenotype is variable, from severe expression [16] with isolated clitoral hypertrophy to moderate expression with hypospadias or isolated micropenis [17]. Adrenal insufficiency may be associated but is not systematically observed [18].
In some patients, the gonadal dysgenesis is associated with renal dysfunction. In these cases, the diagnosis of Drash syndrome—defined as Wilms tumor associated with renal insufficiency—or Frasier syndrome—which is proteinuria secondary to focal glomerular sclerosis—may be made. Both syndromes are due to WT1 gene abnormalities that are nevertheless quite specific for each syndrome. In particular, heterozygous mutations in the open reading frame have been associated with Drash syndrome [19], while intron mutations leading to splicing abnormalities have been found in Frasier patients [20].
Sox9 gene abnormalities have been reported. Sox9 is a key gene in early male sex determination [15]. Several mutations have been identified in patients with severe skeletal malformations like campomelic dysplasia, associated in some cases with sex reversal and gonadal dysgenesis [21–23].
Homozygous or composite heterozygous mutations of the desert hedgehog (DHH) gene, which is involved in testis differentiation and perineal development, have been identified. The virilization defect is frequently severe, the phenotype is often female, and a neuropathy may be associated.
A linkage study recently identified an MAP3K1 mutation in two families with several cases of 46,XY DSD, thus indicating another player in male sex determination [24].
In addition, duplications in the short arm of the X chromosome [dosage sensitive sex reversal (DSS) locus, DAX1 gene] have been reported in several cases of gonadal dysgenesis. A DSS locus duplication was found in 46,XY DSD patients with female phenotype (46,XY complete dysgenesis). Both the DAX1 (DSS-AHC critical region on X chromosome, gene 1) and NR0B1 (nuclear receptor 0B1) genes have been identified in this locus. The NR0B1 gene belongs to the nuclear receptor family and, through its linkage with other transcription factors such as SF1, it has an “anti-testis” effect during the process of male sex determination, proportional to the gene dosage. Thus, the overexpression of the DAX gene, as in the case of duplication in a 46,XY patient, may contrast with normal testis differentiation, leading to gonadal dysgenesis [25].
Last, a chromobox homolog 2 (CBX2) gene mutation was recently identified in a newborn with complete female phenotype but a 46,XY karyotype, which had been determined in the prenatal period because of concerns about maternal age. This testis differentiation abnormality nevertheless differed from the previously presented cases of gonadal dysgenesis in that ovaries with primordial follicles were detected [26]. A mutation in the CBX2 gene, which is known for activating SF1 transcription, was identified in this patient. The CBX2 gene thus seems to actively repress ovarian development in 46,XY gonads.
2.2.2.2 Defects in Testosterone Production
Defects in T production are rare and are characterized by variable degrees of external genital undervirilization. Conversely, no Mullerian derivatives are present because AMH is normally secreted by the Sertoli cells. These defects are due to an enzymatic defect in T biosynthesis or they may be secondary to an LH receptor gene abnormality.
Defect in 3-Beta-Hydroxysteroid Dehydrogenase
This defect is associated with a variable but insufficient virilization of the 46,XY fetus, ranging from a female phenotype to minor forms of DSD, such as isolated micropenis or salt-wasting conditions. The biological and genetic investigations are the same as for 46,XX DSD.
Defect of 17-Alpha-Hydroxylase
The phenotype in cases of a 17-alpha-hydroxylase defect may also be extremely variable. In some individuals, the diagnosis is made only in the pubertal period because of pubertal delay or stagnation associated with gynecomastia. A DOC excess causes hypertension during puberty. The plasma levels of pregnenolone, progesterone, and corticosterone are elevated, which contrasts with the low values of T and D4 that are unresponsive to stimulation. The genetic abnormality concerns the CYP17 gene with recessive transmission.
Defect of 17-Beta-Hydroxysteroid Reductase
This is a rare testicular block that causes a deficit in testicular T production. The phenotype is more frequently female at birth. The diagnosis is based on a striking elevation in plasma D4 contrasting with a low T level. The mutation involves the 17ß-HSD type 3 gene, which is expressed only in testis, and its transmission is recessive. Virilization occurs at puberty associated with gynecomastia.
P450-Oxydoreductase (POR) Deficit
A POR deficit may cause 46,XY DSD as well as 46,XX DSD [7]. The cytochrome P450 oxydoreductase protein enables the electron transport from NADPH to P450 cytochromes localized in microsomes. Several cytochromes take part in cholesterol biosynthesis, while three are involved in steroid biosynthesis: P450C17 (17a-hydroxylase/17,20 lyase), P450C21 (21-hydroxylase), and P450CYP19 (aromatase). Transmission of POR gene mutations is autosomal recessive, and the study of several of these mutations has provided greater insight into DSD in association with combined deficits in 21-OH and 17-OH in cases where molecular analysis of CYP21 and CYP17 was normal. In addition to DSD, several patients present craniofacial malformations, suggestive of Antley-Bixler syndrome. The great variability in the phenotype and endocrine findings makes this diagnosis very difficult. This may be due in part to the varying degrees of the enzymatic defects and to the differences in the ability of each mutation to alter enzyme function [27].
Leydig Cell Agenesis or Hypoplasia
This is a rare form of 46,XY DSD, first identified in a patient with female phenotype associated with the 46,XY karyotype. She presented primary amenorrhea and no breast development at puberty, associated with low T at baseline and after hCG stimulation testing. The discrepancy between increased LH and normal FSH levels is generally evocative. This condition is determined by a homozygous or double heterozygous inactivating mutation of the LH receptor gene. Since the identification of these genetic abnormalities, the phenotypic expression has expanded to include conditions that range from ambiguous genitalia to partial forms such as hypospadias or isolated micropenis [28].
Related posts:
 Central Precocious Puberty: From Diagnosis to Treatment
Amenorrhoea and Anorexia Nervosa in Adolescent Girls
Premature Ovarian Failure in Adolescence and Young Adults: From Diagnosis to Therapy and Follow-up for Fertility Preservation
Emergency Contraception
Central Precocious Puberty: From Diagnosis to Treatment
Amenorrhoea and Anorexia Nervosa in Adolescent Girls
Premature Ovarian Failure in Adolescence and Young Adults: From Diagnosis to Therapy and Follow-up for Fertility Preservation
Emergency Contraception
 Delayed Puberty: Impact on Female Fertility
Delayed Puberty: Impact on Female Fertility
 Adolescent Pregnancy and Contraception
Adolescent Pregnancy and Contraception
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


