Table 32.1 Causes of hypogonadotrophic hypogonadism in males
| Genetic |
| Kallman’s syndrome |
| GnRH receptor mutations |
| Constitutional delay of puberty |
| Hypothalamic |
| Tumours: craniopharyngioma, dysgerminoma |
| Infiltration: sarcoidosis, histiocytosis |
| Postradiotherapy or chemotherapy |
| Pituitary |
| Tumours |
| Infarction or haemorrhage |
| Trauma |
| Head injuries |
| Functional |
| Weight loss |
| Systemic illness |
| Idiopathic |
| Idiopathic hypogonadotrophic hypogonadism |
| Drugs |
| Anabolic steroids |
| Others (rare) |
| Prader–Willi syndrome |
| Laurence–Moon–Biedl syndrome |
| Congenital adrenal hyperplasia |
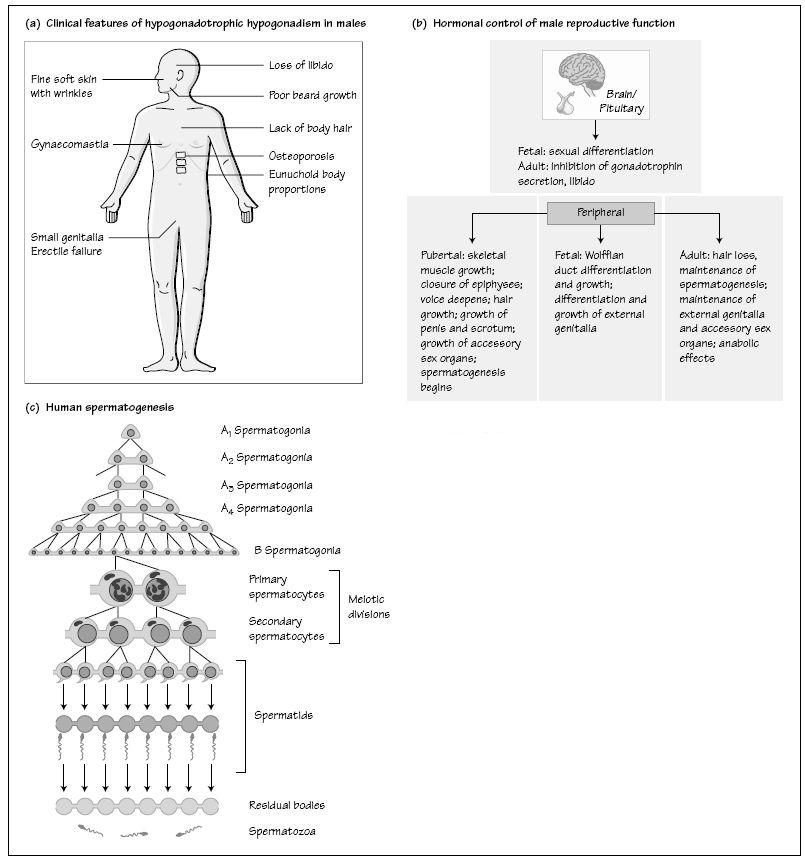
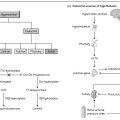
Effects of the failure of androgen action may be best seen in patients with hypogonadotrophic hypogonadism (Fig. 32a). This is caused by a failure of hypothalamic GnRH secretion or by pituitary disease resulting in impaired gonadotrophin release and hence low androgen concentrations (Table 32.1). The clinical features of hypogonadotrophic hypogonadism depend on the timing of its onset, such that males developing the condition after puberty present with features of secondary testicular failure (poor libido, loss of secondary sexual characteristics and subfertility). Prior to puberty, boys present with delayed or failed puberty or, less commonly, the condition presents in the neonatal period with cryptorchidism and micropenis Idiopathic hypogonadotrophic hypogonadism describes those patients in whom there are no anatomical abnormalities of the hypothalamus and pituitary and no associated endocrine disorders.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


