- 6.1 Introduction
- 6.2 Biology of malignant transformation in haematopoietic cells
- 6.3 Leukaemias
- 6.3.1 Acute lymphocytic leukaemia
- 6.3.2 Chronic lymphocytic leukaemia
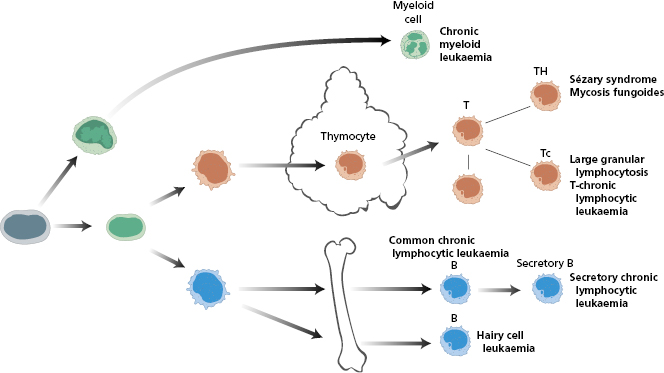
- 6.3.3 Other chronic lymphoid leukaemias
- 6.3.1 Acute lymphocytic leukaemia
- 6.4 Lymphomas
- 6.4.1 Hodgkin’s disease
- 6.4.2 Non-Hodgkin’s lymphoma
- 6.4.1 Hodgkin’s disease
- 6.5 Plasma cell dyscrasias
- 6.5.1 Benign paraproteinaemia or monoclonal gammopathy of unknown significance
- 6.5.2 Multiple myeloma
- 6.5.3 Waldenström’s macroglobulinaemia
- 6.5.4 Other plasma cell dyscrasias
- 6.5.1 Benign paraproteinaemia or monoclonal gammopathy of unknown significance
 Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
6.1 Introduction
The cells involved in immune responses undergo ‘reactive’ polyclonal proliferation as a normal response to infection or inflammation. However, monoclonal expansion can give rise to leukaemias, lymphomas or myeloma.
Leukaemia is the malignant proliferation of haematopoietic stem/progenitor cells and may involve cells of lymphoid, myeloid or monocytic lineages. The clone of malignant cells originates in the bone marrow. Leukaemic ‘blasts’ or other less obvious but still malignant cells may then be seen circulating in peripheral blood and may infiltrate other organs such as lymph nodes and the central nervous system.
Tumours of lymphoid cells originating in peripheral lymph tissue constitute the lymphomas. Dissemination of these malignant cells may result in infiltration of other organs including spleen, liver, brain, bone marrow or lungs. The classification of lymphomas is complex. It is important to distinguish different types of lymphoma in order (i) to provide a reliable diagnosis and prognosis for a given patient, and hence (ii) to choose the most effective form of therapy.
Each leukaemia or lymphoma is thought to arise from a single cell, perhaps a stem cell, which undergoes a malignant transformation that enables uncontrolled division of this cell into a ‘clone’ of identical cells. Mutations in many genes that are involved in normal differentiation from stem cell to mature blood cell have been shown to be present in leukaemias and lymphomas. Many classifications have evolved to take into account such molecular abnormalities, although the leukaemia cell morphology, molecular genetics lineage-specific markers that are identified by flow cytometry remain the keystones of diagnosis.
The immunological techniques that can be used to identify the phenotype of the malignant clone and to classify these lymphoid malignancies are shown in Table 6.1. These techniques are discussed fully in Chapter 19. Molecular genetics to determine translocations and chromosomal abnormalities are now used routinely for prognosis.
Table 6.1 Techniques used to identify lymphoid malignancies*
1 Morphology – how the cell and its nucleus look by light microscopy 2 Special cytochemical stains – to identify characteristic surface markers, enzymes, carbohydrates or lipids 3 Immunophenotyping – use of monoclonal antibodies (MAbs) to identify cell lineage (myeloid/B or T lymphocytic) and stage of differentiation by surface and intracellular antigens using flow cytometry 4 Cytogenetic analysis – to identify characteristic abnormal translocations and deletions on chromosomes by visualization, banding and molecular genetics 5 Gene rearrangement studies – can be used to identify or confirm monoclonality, in T-cell malignancies |
*Not all these techniques are needed for every diagnosis of lymphoid malignancy.
6.2 Biology of malignant transformation in haematopoietic cells
In most types of leukaemia and lymphoma, as for solid tumours, the precise stimulus for initiation of the malignant clone is not known. The majority of tumours in man arise spontaneously. Inherited genetic mutations have been identified for a minority of cancers, for example BRCA genes in breast cancer. However, most cancers involve acquired gene mutations (Fig. 6.1). Such mutations commonly arise in genes responsible for cell proliferation, apoptosis (programmed cell death) or DNA repair. Viruses known to be involved in the pathogenesis of lymphoid tumours include the Epstein–Barr virus (EBV) in Burkitt’s lymphoma and the retrovirus human T-cell leukaemia virus I (HTLV-I) in adult T-cell leukaemia/lymphoma. It is believed that in neither case does infection alone cause the tumour, since only 1% or less of infected individuals in endemic areas develop the malignancy. Additional instigating factors are needed, which may include other genetic abnormalities, such as DNA repair gene defects as in ataxia telangiectasia, or environmental factors, such as radiation or infection with other microorganisms.
Fig. 6.1 A malignant translocation: Abl is an oncogene on chromosome 9. Bcr is a similar oncogene on chromosome 22. When these two are joined by translocation between the two chromosomes, activation of the fused gene produces a new tyrosine kinase, which enables uncontrolled proliferation of the cells. This translocation is visualized as the Philadelphia chromosome in almost all patients with chronic myeloid leukaemia and some with acute lymphoblastic leukaemia. Axford J & O′Callaghan C (eds) Medicine, 2e (2004). Reproduced with permission of John Wiley & Sons Ltd.
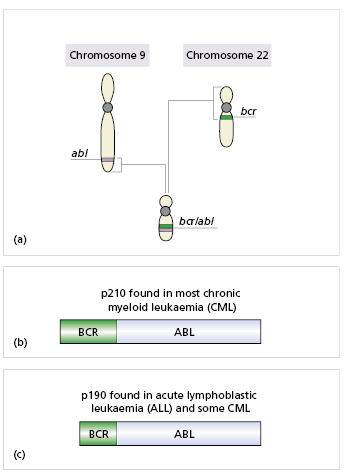
Proto-oncogenes (see Box 6.1) are genes that encode proteins involved in cell growth, differentiation and division. Mutation of these genes to oncogenes can lead to malignancy. Such mutations can result in constitutively (permanently) active proteins, proteins with abnormal function or increased protein concentrations resulting in heightened activity. An example of a translocation resulting in a new cell proliferation enzyme is given in Fig. 6.1. Tumour suppressor genes regulate cell proliferation, DNA repair and apoptosis. Failure of these genes is involved in tumours in general.
- Activation of a positive regulator gene, if the DNA is inserted next to a promoter gene, e.g. ‘insertional mutagenesis’
- Inactivation of a negative gene if the DNA is inserted next to an apoptosis or apoptosis promoter gene, e.g. ‘insertional mutagenesis of apoptosis’
- Activation of a growth factor production gene if the DNA is inserted next to a growth factor gene, resulting in unregulated cell division
- Inactivation of a DNA repair gene if the DNA is inserted next to a DNA repair gene or promoter gene
- Inactivation of a differentiation gene if the DNA is inserted next to a differentiation gene or promoter gene
- Recombination between host cell genes and viral DNA can result in host genes becoming replicating and therefore oncogenic
- Silent ‘proto-oncogenes’ (c-onc genes), incorporated into mammalian cells eons ago, are inherited in a Mendelian fashion, and normally used for growth factor receptors or signal transducers
these can be activated by mutagenesis
- Silent ‘proto-oncogenes’ (c-onc genes), incorporated into mammalian cells eons ago, are inherited in a Mendelian fashion, and normally used for growth factor receptors or signal transducers
- Loss of function if tumour suppression gene inactivated
6.3 Leukaemia
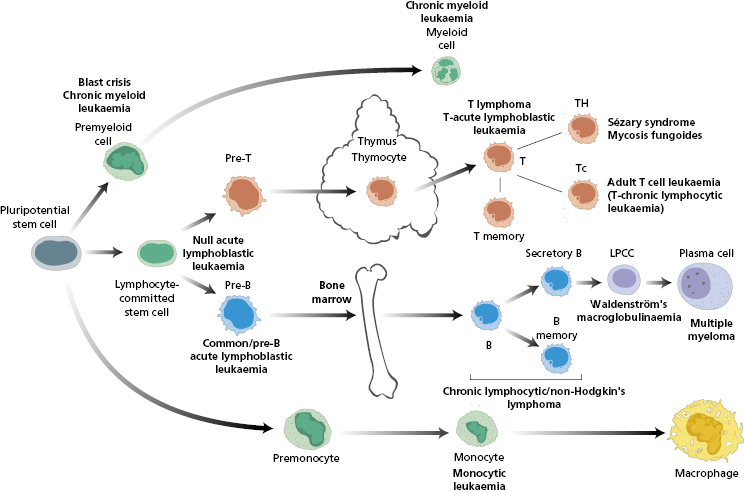
Leukaemias may be lymphoid, myeloid, monocytic or megakaryocytic in origin (Fig. 6.2). The cells may retain some of their original characteristics; however, some are poorly differentiated and the precise lineage can be difficult to identify by morphology alone. This chapter focuses on lymphoblastic leukaemias – both acute lymphoblastic leukaemia (ALL) and chronic lymphocytic leukaemia (CLL); readers are referred to the Essentials of Haematology edition for details on other leukaemias.
Fig. 6.2 Malignant counterparts at each step in the leucocyte differentiation pathway. LPCC, Lymphoplasma cytoid cell; T, T cell; TH, T helper cell; Tc, T cytotoxic cell; B, B cell.
6.3.1 Acute lymphoblastic leukaemia
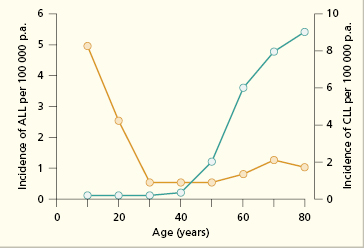
ALL is largely a disease of children and young people; it is less common over the age of 20 years (Fig. 6.3). It is five times more common than acute myeloid leukaemia (AML). Patients with ALL often present with complications of bone marrow failure, including bleeding, infection and anaemia (as in Case 6.1). Other signs/symptoms include sweats and meningeal irritation. Patients may have palpable lymphadenopathy and a small proportion (10%) have a mediastinal mass apparent on chest X-ray. Over 80% of patients are thrombocytopenic and in some this is severe and results in petechiae. Many (60%) have low haemoglobin levels below 80 g/L. The white cell count is usually low (leucopenia) but a minority of patients present with an apparently high white cell count due to circulating blasts and this indicates a poorer prognosis.
Fig. 6.3 Decade of onset in acute lymphoblastic leukaemia (ALL) (yellow line) and chronic lymphocytic leukaemia (CLL) (green line).
 Case 6.1 Acute leukaemia (common type)
Case 6.1 Acute leukaemia (common type)A 7-year-old boy presented with malaise and lethargy of 6 days duration. He had become inattentive at school, anorexic and had lost 3 kg in weight. On examination he was thin, anxious and clinically anaemic. There was mild, bilateral, cervical lymphadenopathy and moderate splenomegaly.
On investigation, he was pancytopenic with a low haemoglobin (80 g/l), platelet count (30 × 109/l) and white cell count (1.2 × 109/l). The blood film showed that most leucocytes were blasts; the red cells were normochromic and normocytic. Bone marrow examination showed an overgrowth of primitive white cells with diminished numbers of normal erythroid and myeloid precursors. Acute leukaemia was diagnosed.
The circulating blast cells were typed by immunological methods: they did not react with monoclonal antibodies to human T-cell precursor antigens (CD2, CD7), but they were positive for major histocompatibility complex class II (DR), common acute lymphoblastic leukaemia (CD10) and B-cell precursor (CD19) antigens, and contained the enzyme terminal deoxynucleotidyl transferase (Tdt) (see Table 6.2). The phenotype of the blasts was that of acute leukaemia of early precursor B cells (see Fig. 6.2), and the prognosis in this child was relatively good. Cytogenetics confirmed ETV6-RUNX1 mutation or t(12;21) translocation, indicating an good prognosis.
The diagnosis of leukaemia is confirmed by performing a bone marrow aspirate and trephine biopsy and finding that >20% of cells are leukaemic blast cells (see Case 6.1). The blasts in the marrow and peripheral blood are immunophenotyped using flow cytometry (Table 6.2) and immunohistology.

ALL may be of B- or T-cell origin. In B-ALL, the malignant cells have the B-cell surface markers, CD10+, CD19+, CD24+ and cytoplasmic CD79+ as well as terminal deoxynucleotidyl transferase (TdT). There is variable expression of lymphoid lineage CD45, and confusingly some cells may have aberrant expression of myeloid lineage surface markers such as CD13 and CD33.
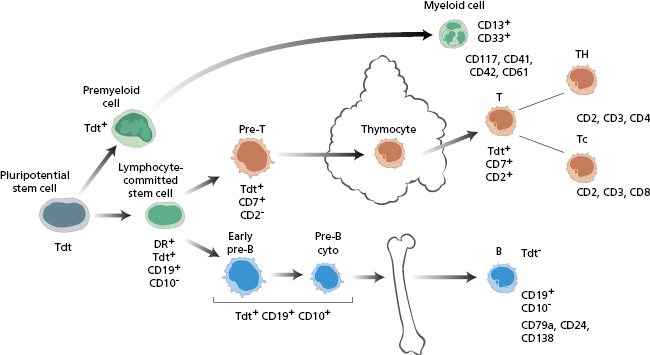
The malignant cells in T-ALL express T-cell markers such as CD2, CD3, CD4, CD5, CD7 and CD8, reflecting transformation of intrathymic T cell development (Fig. 6.4).
Fig. 6.4 Markers of various forms of acute lymphoblastic leukaemia (ALL). See Fig. 6.2 for abbreviations.
In addition to flow cytometry and histology, cytogenetic studies are performed to identify genetic mutations that correlate with prognosis. For example, in B-ALL, poor prognostic indicators include presence of the BCR-ABL fusion protein caused by the t (9;22) translocation and rearrangements of the MLL gene. The TEL-AML1 or t (12;21) translocation is associated with a good prognosis. Chromosomal translocations involving T-cell receptors are seen in about a third of T-ALL cases; these include NOTCH1, FBW7, MYB, LMO1/2 and CALM-AF10 as well as TCR γ/δ. Better prognosis in T-ALL patients occurs with some HOX rearrangements and some TAL1/SCL mutations.
Other factors used as prognostic criteria in ALL include the white cell count and age at presentation. Children under 1 year old or over 10 years and those with a high blast cell count at presentation (>50 × 109/L) are considered high risk.
Complications of ALL include anaemia and thrombocytopenia, resulting in bleeding due to marrow infiltration (Table 6.3). Concentrated red cells are given to correct anaemia, and platelet concentrates to prevent and treat bleeding. Infection is a common complication of ALL and preventive measures, such as barrier nursing and prophylactic antifungal and antiviral agents, are important.
Table 6.3 Complications of acute lymphoblastic leukaemia (ALL) associated with disease or its treatment with chemotherapy and HSCT transplantation
| Due to disease | Due to treatment |
|---|---|
| Anaemia – bone marrow suppression or hypersplenism Infection – neutropenia due to bone marrow suppression Bleeding – low platelets | Infection – poor neutrophil function and severe neutropenia as a result of chemotherapy Low platelets as result of chemotherapy Graft-versus-host disease secondary to allogeneic transplant |
| Leukaemic infiltrates, e.g. meninges, testis, skin | Poor growth Intellectual impairment (especially if cranial irradiation is performed in patients with CNS disease) Cardiac dysfunction |
ALL is a fatal condition if untreated, but aggressive chemotherapy may eliminate the clone of malignant cells and induce a cure (see Box 6.2). B-ALL has an excellent outcome in children. A complete remission is achieved with initial high-dose ‘induction’ chemotherapy in around 95% of children and 60–85% for adults. Further treatment is then required to ‘consolidate’ and ‘maintain’ the remission. The intensity of the consolidation therapy depends on prognostic factors at presentation and on the initial response to chemotherapy, in particular whether there is any residual disease following induction chemotherapy. For higher risk patients, an allogeneic human stem cell transplant [HSCT] may be performed if a suitably matched donor is identified.
There are complications associated with therapy, both with initial chemotherapy and HSCT (Table 6.3). Severe infection secondary to the profound neutropenia that results from intensive therapy is the major cause of mortality and morbidity in ALL. Barrier nursing and prompt and aggressive treatment of neutropenic fever according to standardized protocols can reduce the risk of death from sepsis in neutropenic patients, similar to the protocols used after human stem cell transplantation for primary immunodeficiencies (see Chapter 3 for Immunodeficiencies and Chapter 8 for Transplantation).
Graft-versus-host disease (GvHD) may occur after allogeneic stem cell transplantation, although some graft-versus-leukaemia effect is helpful for the anti-leukaemic effect. Long-term complications of survivors such as organ damage, infertility and psychological issues must also be considered.
6.3.2 Chronic lymphocytic leukaemia
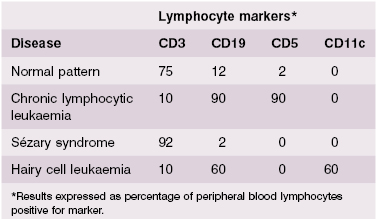
Chronic lymphocytic leukaemia (CLL) is a relatively common disease of elderly patients (20 per 105 prevalence in those >60 years old). It is uncommon in people under 50 years of age (see Fig. 6.3), and usually runs a relatively benign course, although speed of progression varies enormously. Excessive numbers of small lymphocytes are found in the peripheral blood (Fig. 6.5); in over 90% of cases of CLL, the neoplastic cells are B-cells (Fig. 6.6). They have the characteristic cell surface markers of circulating B cells (Case 6.2 and Table 6.4). The cells represent a malignant proliferation of a single clone of B cell and are therefore, as in acute leukaemias, ‘monoclonal’. Normally ‘reactive’ lymphocytic proliferation is polyclonal and therefore the ratio of cells with κ or λ is 3 : 2. In contrast, in a monoclonal B-cell proliferation, this ratio is changed in favour of the malignant clone as all the malignant cells express the same light-chain type. These cells accumulate progressively in the blood, lymph nodes, spleen, liver and bone marrow.
 Case 6.2 Chronic lymphocytic leukaemia
Case 6.2 Chronic lymphocytic leukaemiaA 62-year-old man presented with general lethargy, night sweats and loss of weight. He had suffered five chest infections during the previous winter, despite being a non-smoker. On examination, there was moderate, bilateral cervical lymphadenopathy and left inguinal lymph node enlargement. The spleen and liver were enlarged 5 cm below the costal margins. There was no bone tenderness and there were no lesions in the skin. On investigation, his haemoglobin was slightly low, (10.2 g/l) the platelet count (251 × 109/l) was normal but his white cell count was increased to 150 × 109/l; the film showed that 98% of these were small lymphocytes.
The features on the blood film were suggestive of chronic lymphocytic leukaemia and immunophenotyping confirmed this diagnosis. Ninety per cent of the cells were B cells (CD 19+); they all expressed CD 19 and CD5 confirming the diagnosis (Table 6.4). The serum immunoglobulins were low: IgG 2.2 g/l (NR 7.2–19.0 g/l); IgA 0.6 g/l (NR 0.8–5.0 g/l) and IgM 0.4 g/l (NR 0.5–2.0 g/l). There was no monoclonal immunoglobulin in the serum or the urine.
Fig. 6.5 CLL blood film. Axford J & O′Callaghan C (Eds) Medicine, 2nd Ed (2004). Reproduced with permission of John Wiley & Sons Ltd.
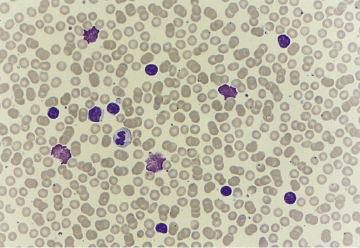
Although most elderly patients with CLL usually have a relatively benign illness and survive for over 8–10 years, the prognosis is variable. Attempts are ongoing to develop a protocol combining clinical and molecular characteristics to provide prognostic information; genomic analyses have already identified some genes, for example p53
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


