The molecular characterization of lung cancer has changed the classification and treatment of these tumors, becoming an essential component of pathologic diagnosis and oncologic therapy decisions. Through the recognition of novel biomarkers, such as epidermal growth factor receptor mutations and anaplastic lymphoma kinase translocations, it is possible to identify subsets of patients who benefit from targeted molecular therapies. The success of targeted anticancer therapies and new immunotherapy approaches has created a new paradigm of personalized therapy and has led to accelerated development of new drugs for lung cancer treatment. This article focuses on clinically relevant cancer biomarkers as targets for therapy and potential new targets for drug development.
Key points
- •
The molecular characterization of lung cancer has changed the classification and treatment of these tumors, becoming an essential component of pathologic diagnosis and therapy decisions.
- •
The success of targeted therapies and new immunotherapy approaches has created a new paradigm of personalized therapy in lung cancer.
- •
Pathologists should be able to precisely handle tissue adequacy in terms of quantity and quality and maintaining tumor cells for detection of molecular alterations.
- •
This article focuses on clinically relevant cancer biomarkers as targets for therapy, and potential new targets for drug development.
Introduction
Lung cancer has shown a decrease in incidence and mortality in recent decades; however, it remains one of the cancers with the highest incidence and ranks first in cancer-related deaths in the United States. An estimated 221,200 new cases and 158,040 deaths are expected to occur in 2015, representing approximately 13% of all cancers diagnosed and 27% of all cancer deaths. Despite advances in early detection and standard treatment, most patients are diagnosed at an advanced stage and have a poor prognosis, with an overall 5-year survival rate of 10% to 15%. Lung cancer is a heterogeneous disease comprising several subtypes with pathologic and clinical relevance. The recognition of histologic subtypes of non–small cell lung carcinoma (NSCLC), namely adenocarcinoma, squamous cell carcinoma, and large cell lung carcinoma as the most frequent subtypes, has become important as a determinant of therapy in this disease. In addition, in recent years, the identification of molecular abnormalities in a large proportion of patients with lung cancer has allowed the emergence of personalized targeted therapies and has opened new horizons and created new expectations for these patients. The use of predictive biomarkers to identify tumors that could respond to targeted therapies has meant a change in the paradigm of lung cancer diagnosis.
This paradigm change affects all stakeholders in the fight against lung cancer including pathologists. Currently, several multiplex genotyping platforms for the detection of oncogene mutations, gene amplifications, and rearrangement are moving to the clinical setting. Genome-wide molecular investigations using next-generation sequencing (NGS) technologies have been evaluated in the research setting, with promising results. Further investigations in NSCLC are required for a better understanding of the implications of intratumor heterogeneity and the roles of tumor suppressor genes and epigenetic events with no known driver mutations. NGS in the clinical setting will provide comprehensive information cheaper and faster by using small amounts of tissue. Pathologists should be able to precisely handle tissue adequacy in terms of quantity and quality and maintaining tumor cells for detection of molecular alterations. The recent clinical successes of immunotherapy approaches to lung cancer have posed additional challenges to the scientific community and pathologists to develop predictive biomarkers of response to these therapies and have highlighted the need for proper procurement and processing of tissue specimens from patients with lung cancer.
This article focuses on the major predictive biomarkers in NSCLC, with special emphasis on their clinical and molecular importance, and the current status of molecular testing for these biomarkers.
Introduction
Lung cancer has shown a decrease in incidence and mortality in recent decades; however, it remains one of the cancers with the highest incidence and ranks first in cancer-related deaths in the United States. An estimated 221,200 new cases and 158,040 deaths are expected to occur in 2015, representing approximately 13% of all cancers diagnosed and 27% of all cancer deaths. Despite advances in early detection and standard treatment, most patients are diagnosed at an advanced stage and have a poor prognosis, with an overall 5-year survival rate of 10% to 15%. Lung cancer is a heterogeneous disease comprising several subtypes with pathologic and clinical relevance. The recognition of histologic subtypes of non–small cell lung carcinoma (NSCLC), namely adenocarcinoma, squamous cell carcinoma, and large cell lung carcinoma as the most frequent subtypes, has become important as a determinant of therapy in this disease. In addition, in recent years, the identification of molecular abnormalities in a large proportion of patients with lung cancer has allowed the emergence of personalized targeted therapies and has opened new horizons and created new expectations for these patients. The use of predictive biomarkers to identify tumors that could respond to targeted therapies has meant a change in the paradigm of lung cancer diagnosis.
This paradigm change affects all stakeholders in the fight against lung cancer including pathologists. Currently, several multiplex genotyping platforms for the detection of oncogene mutations, gene amplifications, and rearrangement are moving to the clinical setting. Genome-wide molecular investigations using next-generation sequencing (NGS) technologies have been evaluated in the research setting, with promising results. Further investigations in NSCLC are required for a better understanding of the implications of intratumor heterogeneity and the roles of tumor suppressor genes and epigenetic events with no known driver mutations. NGS in the clinical setting will provide comprehensive information cheaper and faster by using small amounts of tissue. Pathologists should be able to precisely handle tissue adequacy in terms of quantity and quality and maintaining tumor cells for detection of molecular alterations. The recent clinical successes of immunotherapy approaches to lung cancer have posed additional challenges to the scientific community and pathologists to develop predictive biomarkers of response to these therapies and have highlighted the need for proper procurement and processing of tissue specimens from patients with lung cancer.
This article focuses on the major predictive biomarkers in NSCLC, with special emphasis on their clinical and molecular importance, and the current status of molecular testing for these biomarkers.
Histologic subtyping of non–small cell lung carcinoma
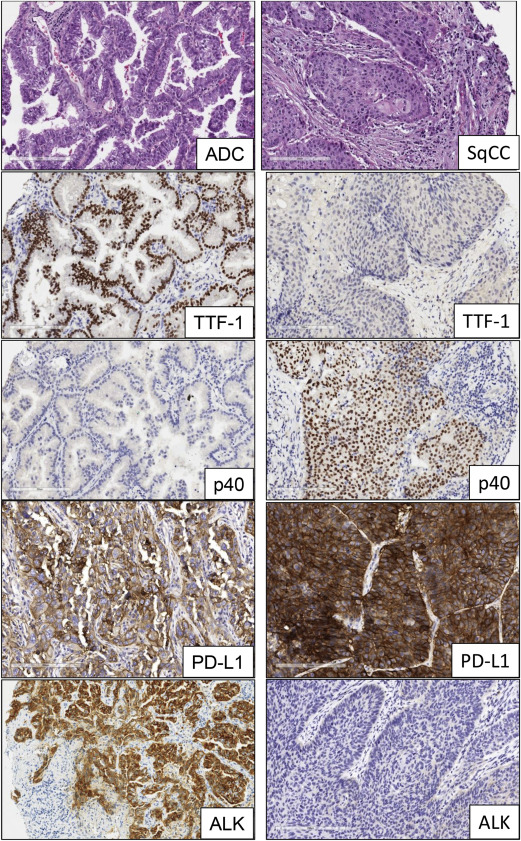
The advent of molecular profiling and targeted therapy has renewed interest in the classification of NSCLC into major subtypes, such as adenocarcinoma, squamous cell carcinoma, and large cell lung carcinoma. Other subtypes, including sarcomatoid carcinoma and neuroendocrine large cell carcinoma, represent a very minor proportion of the total NSCLC cases. The most recent histologic classification of lung cancer published by the World Health Organization in 2015 incorporates relevant genetics and immunohistochemistry (IHC) aspects of different tumor subtypes ( Fig. 1 ). Lung cancers are increasingly diagnosed and staged by transthoracic core needle biopsy and fine-needle aspiration, transbronchial needle aspiration, endobronchial ultrasound-guided transbronchial needle aspiration, and endoscopic ultrasound-guided fine-needle aspiration. It is well established that poorly differentiated adenocarcinoma and squamous cell carcinoma of the lung can appear indistinguishable by routine microscopy, particularly in small biopsy and cytology specimens. In these small specimens, particularly in poorly differentiated tumors, there is a need to integrate morphology with IHC analysis to make a precise diagnosis. This includes the examination of IHC expression of thyroid transcription factor and the novel aspartic proteinase of the pepsin family A (napsin A) for adenocarcinoma and p40 and cytokeratin 5/6 for squamous cell carcinoma. In addition, histochemical staining of mucin is useful for the diagnosis of adenocarcinoma histology. The correct histologic diagnosis of these specimens is important, but it is also imperative to exercise judicial use of the tissue to maximize the yield for molecular testing ( Table 1 ).
| Gene | Alteration | Adenocarcinoma Frequency, % | Squamous Cell Carcinoma Frequency, % |
|---|---|---|---|
| EGFR | Mutation | 10 | 3 |
| ALK | Rearrangement | 4–7 | None |
| ROS | Rearrangement | 1–2 | None |
| KRAS | Mutation | 25–35 | 5 |
| MET | Mutation | 8 | 3 |
| MET | Amplification | 4 | 1 |
| NTRK1 | Rearrangement | 3 | None |
| FGFR | Amplification | 3 | 20 |
| HER2 | Mutation | 1.6–4 | None |
| BRAF | Mutation | 1–3 | 0.3 |
| PIK3CA | Mutation | 2 | 7 |
| RET | Rearrangement | 1–2 | None |
| DDR2 | Mutation | 0.5 | 3–4 |
| PTEN | Deletion | — | 16 |
Genomic biomarkers in non–small cell lung carcinoma
Advances in elucidating the molecular biology of lung cancer have led to the identification of several potential biomarkers that could be relevant in the clinical management of patients with NSCLC.
Epidermal Growth Factor Receptor
The epidermal growth factor receptor (EGFR) is a tyrosine kinase receptor member of the ERBB family. The EGFR gene is located on the short arm of chromosome 7 at position 12. When the extracellular ligand binds to EGFR, it generates homodimerization or heterodimerization of the receptor, leading to phosphorylation of sites in the cytoplasmic tyrosine kinase and activation of various intracellular pathways, including the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) and RAS/RAF/mitogen-activated protein kinase (MAPK) pathways, which lead to cell proliferation, metastasis, and prevention of apoptosis. EGFR is overexpressed in 62% of NSCLCs, and its expression has been associated with poor prognosis. Approximately 10% of patients with adenocarcinoma of the lung in the United States and 30% to 50% in East Asia have lung tumors associated with EGFR mutations. These mutations occur within exons 18 to 21, which encode for a portion of the EGFR kinase domain. Approximately 90% of EGFR mutations occur as in-frame deletions in exon 19 or as missense mutations in exon 21 (44% and 41% of all mutations, respectively). Activating mutations in the kinase domain of EGFR trigger ligand-independent tyrosine kinase activation, leading to hyperactivation of downstream antiapoptotic signaling pathways. EGFR mutations are found more often in adenocarcinomas with lepidic features from female never-smokers. The high response rates (55%–78%) to treatment with tyrosine kinase inhibitors (TKIs), such as gefitinib, erlotinib, and afatinib, in patients with EGFR -mutant tumors, and the significantly greater progression-free survival (PFS) of these patients, have made EGFR TKIs the standard treatment of patients with these mutations. However, most of these patients develop resistance and relapse in a short time, because of the occurrence of a new mutation (T790 M) in exon 20 of the EGFR kinase domain (50%), amplification of the MET oncogene (21%), or mutations of PI3KCA .
EGFR mutations are identified mostly with the use of gene sequencing methodologies and real-time polymerase chain reaction (PCR)-based assays. Both methods have been reported to have high performance and sensitivity in the detection of these mutations in formalin-fixed and paraffin-embedded tissues. Detection of EGFR mutations by an IHC-based approach with specific antibodies against mutant proteins has been attempted but showed variable sensitivity and significant variability between studies.
Anaplastic Lymphoma Kinase
Anaplastic lymphoma kinase (ALK) is a tyrosine kinase receptor member of the insulin receptor superfamily. The ALK gene is located on the short arm of chromosome 2 at position 23. ALK gene rearrangement was originally identified in anaplastic large cell lymphoma and was subsequently described in a subset of NSCLC tumors harboring a fusion of ALK and echinoderm microtubule-associated protein-like 4 ( EML4 ) genes. This rearrangement encodes for a chimeric protein with constitutive kinase activity, which promotes malignant growth and proliferation. The EML4-ALK fusion has been detected in 3.7% to 7% of NSCLCs, usually in adenocarcinomas with signet-ring cells or cribriform histology features, and is more common in young patients who have never smoked. There are several EML4-ALK rearrangement variants and also ALK fusion with other less frequent partners, such as kinesin family member 5B ( KIF5B ), TRK-fused gene ( TFG ), kinesin light chain 1 ( KLC1 ), and huntingtin-interacting protein 1 ( HIP1 ) genes, resulting in oncogenic transformation. It has been shown that EGFR , Kirsten rat sarcoma viral oncogene homolog gene ( KRAS ), and ALK molecular alterations are mutually exclusive events ; nevertheless, they have been described in up to 2.7% of lung adenocarcinoma cases with concurrent molecular alterations. The ALK fusion defines a distinct subpopulation of patients with lung adenocarcinoma who are highly responsive (57%–74%) to ALK inhibitors, such as crizotinib. Patients treated with crizotinib demonstrated significantly better median PFS and response rate compared with patients who received chemotherapy. As a result, testing for ALK rearrangements in patients with advanced lung adenocarcinoma is recommended in current clinical practice guidelines. However, despite initial responses, a fraction of the patients develop acquired resistance to crizotinib, because of secondary mutations within the kinase domain of EML4-ALK; these include L1196 M, C1156Y, and F1174 L, among others. Several second-generation ALK inhibitors that target ALK-positive NSCLC, such as alectinib, ceritinib, and AP26133, have been developed and are currently under evaluation in clinical trials.
Current diagnostic approaches to detect ALK fusion genes and their results include break-apart fluorescence in situ hybridization (FISH), IHC, and reverse-transcription PCR (RT-PCR). Break-apart FISH has been established in clinical trials as the standard method for confirmation of ALK status. FISH and IHC have shown high concordance in several reports, especially with the development of IHC antibodies (clones 5A4 and D5F3) with better sensitivities and specificities (83%–100%) for the detection of ALK rearrangement. As a result, IHC detection of the ALK protein is being considered as an adequate screening tool to test NSCLC samples for ALK rearrangements or as a tool to evaluate cases that are not interpretable by FISH. Nevertheless, other studies reveal major discordances, suggesting the need to combine both tests to optimize detection. Other methods, such as RT-PCR and NGS, are in use but have not been examined systematically compared with FISH as a predictor of response to ALK inhibitors.
Kirsten Rat Sarcoma Viral Oncogene Homolog
KRAS is an oncogene located on the long arm of chromosome 12 at position 12.1. It is a member of the RAS family of membrane-associated G proteins and encodes for a protein with intrinsic GTPase activity, which is involved in a variety of cellular responses including proliferation, cytoskeletal reorganization, and survival. KRAS acts downstream of several tyrosine kinases receptors, including EGFR, and is associated with activation of the RAS/RAF/MAP kinase (MEK)/extracellular signal-regulated kinase (ERK) and RAS/MAPK signaling pathways. KRAS mutations occur in 25% to 35% of patients with NSCLC, principally adenocarcinomas with a solid pattern, and are found more often in white patients compared with Asians, in former or current smokers, but without sex predilection. Mutations in the form of single-nucleotide missense variants are found in codons 12 and 13 in approximately 95% of cases. In never-smokers, the most common KRAS mutations are G12D and G12 V, whereas G12 C is the most common mutation associated with smoking. The presence of KRAS mutation may be associated with unfavorable outcome and could be a negative predictor of responsiveness to chemotherapy. In addition, it is associated with an increased likelihood of having a second primary tumor and is a predictor of resistance to targeted therapy with EGFR-TKIs, such as gefitinib or erlotinib, in patients with NSCLC.
Because KRAS , EGFR , and ALK molecular alterations are mutually exclusive, it has been suggested that KRAS testing could be a surrogate assay to exclude EGFR – and ALK -positive cases ; however, this approach is not currently recommended. Although there are no targeted therapies approved for patients with lung cancer and KRAS mutation, several clinical trials aimed at downstream signaling targets are under way. Different phase 2 trials have reported improvements in PFS and response rate with the combination of selumetinib (MEK1/MEK2 inhibitor) and docetaxel compared with docetaxel alone and promising results with sorafenib (RAS/RAF pathway inhibitor), with a disease control rate of approximately 50%. Conversely, trametinib (MEK1/MEK2 inhibitor) did not show advantages over docetaxel in patients with NSCLC.
ROS Proto-oncogene 1, Receptor Tyrosine Kinase
ROS proto-oncogene 1, receptor tyrosine kinase (ROS1) is a tyrosine kinase receptor member of the insulin receptor family and is located on the long arm of chromosome 6 at position 22. ROS1 plays a role in epithelial cell differentiation during the development of a variety of organs, but no ligand for this receptor has been identified. ROS1 rearrangements were originally described in glioblastoma and have also been reported in cholangiocarcinoma and ovarian cancer. Approximately 1% to 2% of NSCLCs harbor ROS1 rearrangements, and several fusion partners including CD74 , solute carrier family 34, member 2 ( SLC34A2 ), leucine-rich repeats and immunoglobulin-like domains 3 ( LRIG3 ), ezrin ( EZR ), syndecan 4 ( SDC4 ), tropomyosin 3 ( TPM3 ), and FIG have been reported in these tumors. All of these fusions result in a chimeric protein that has been reported to be oncogenic. ROS1 -rearranged NSCLC typically occurs in young, female, never-smokers with a histologic diagnosis of adenocarcinoma and is usually mutually exclusive with other oncogenic drivers ( EGFR , KRAS , ALK ). Clinical trials have reported that patients with advanced NSCLC harboring ROS1 rearrangement have benefited from crizotinib treatment, showing response rates up to 80%. Ongoing phase 1 and 2 studies are investigating the activity of crizotinib and ceritinib (ALK inhibitor) in ROS1 -rearranged NSCLC. ROS1 testing is indispensable for identifying patients who could benefit from crizotinib treatment. The National Comprehensive Cancer Network 2014 guidelines recommend that all patients with advanced triple-negative ( EGFR , ALK , and KRAS ) lung adenocarcinoma be tested for other molecular markers including ROS1 .
There is not a gold standard method, but currently available diagnostic methods include FISH, RT-PCR, and IHC. FISH is the only method approved by the US Food and Drug Administration to detect ALK- rearranged NSCLC and has been used in clinical trials as the standard method for confirmation of ROS1 rearrangement; nevertheless, it is an expensive and laborious technique. Because ROS1 -rearranged lung cancer is rare, assessment of ROS1 protein expression by IHC may be used as a screening tool for the identification of candidates suitable for ROS1-targeted therapy. In fact, studies have found that ROS1 IHC (D4D6 clone) has a high sensitivity (100%) and specificity (92%–97%) for ROS1 rearrangements compared with FISH.
Human Epidermal Growth Factor Receptor 2
The human EGFR 2 gene HER2 ( ERBB2 ) is a proto-oncogene located on chromosome 17 at position 12. It encodes for a tyrosine kinase receptor member of the ERBB receptor family. HER2 lacks a specific ligand. Nevertheless, it can be combined with other ERBB receptors to form a heterodimer. This allows for the activation of important signal transduction pathways, including the MAPK and PI3K pathways, involved in cell proliferation, differentiation, and migration. HER2 expression and/or amplification are found in many cancers including breast and gastric cancer. Overexpression of HER2 has been reported in 7% to 34.9% of NSCLCs and has been associated with poor prognosis in patients with these tumors. Activating mutations of HER2 have been found in 1.6% to 4% of lung cancers. These mutations occur in the four exons of the tyrosine kinase domain (exons 18–21) and are found more often in adenocarcinomas in female, Asian, never-smokers, or light smokers. HER2 mutations are almost always mutually exclusive with other driver oncogene alterations in lung cancer described previously. Different studies reinforce the importance of screening lung adenocarcinomas for HER2 mutation as a method to select patients who could benefit from HER2 -targeted therapies (afatinib and trastuzumab), which have shown response rates of approximately 50%. Several clinical trials of targeted agents, such as trastuzumab, neratinib, and pyrotinib, among others, are being conducted in patients with HER2 mutation. HER2 mutations are usually assessed via sequencing approaches.
RET Proto-oncogene
The RET proto-oncogene is located on the long arm of chromosome 10 at position 11.2. It encodes for a tyrosine kinase receptor for the glial cell line–derived neurotrophic factor family of ligands and is involved in cell proliferation, migration, and differentiation, and neuronal navigation. RET chromosomal rearrangements were originally described in papillary thyroid carcinoma. Approximately 1% to 2% of NSCLCs harbor RET fusions, and several fusion partners, including kinesin family member 5B ( KIF5B ; 90%), coiled-coil domain containing 6 ( CCDC6 ), nuclear receptor coactivator 4 ( NCOA4 ), and tripartite motif-containing 33 ( TRIM33 ), have been described. RET -rearranged NSCLC typically occurs in adenocarcinomas with more poorly differentiated solid features in young never-smokers, and it is mutually exclusive with known driver oncogenes. In vitro studies showed that RET fusions lead to oncogenic transformation, which can be inhibited by multitargeted kinase inhibitors, such as vandetanib, sorafenib, and sunitinib. Preliminary studies with cabozantinib (MET and vascular endothelial growth factor receptor 2 inhibitor) in RET -rearranged lung adenocarcinoma are promising.
FISH is currently the standard diagnostic assay for detection of RET chromosomal rearrangements. RT-PCR is usually insufficient for the detection of new partners or isoforms, and RET IHC has shown low sensitivity and specificity for RET rearrangements. Sequencing approaches, including NGS methodologies, are also frequently used to detect RET translocations.
MET Proto-oncogene
The MET gene is located on the long arm of chromosome 7 at position 31. This oncogene encodes for a tyrosine kinase receptor (hepatocyte growth factor receptor), which activates multiple signaling pathways that play fundamental roles in cell proliferation, survival, motility, and invasion. Pathologic activation of MET includes mutation, gene amplification, and protein overexpression. MET alterations were first reported in patients with renal papillary carcinoma and mutations in the MET kinase domain leading to constitutive activation of the receptor. In lung cancer, MET mutations are found in the extracellular semaphorin and juxtamembrane domains, occurring in 3% of squamous cell lung cancers and 8% of lung adenocarcinomas. MET amplifications are found in 4% of lung adenocarcinomas and 1% of squamous cell lung cancers and are associated with sensitivity to MET inhibitors. In NSCLC, MET and hepatocyte growth factor protein expression, along with high MET gene copy number, have been described as poor prognosis factors. Activating point mutations affecting splice sites of exon 14 of the MET gene ( MET ex14), which occur in 4% of lung adenocarcinomas, represent a possible oncogenic driver and identify a subset of patients who may benefit from MET inhibitors, such as capmatinib and crizotinib. This novel alteration is usually assayed by NGS methodology.
B-RAF Proto-oncogene, Serine/Threonine Kinase
The B-RAF proto-oncogene, serine/threonine kinase ( BRAF ) oncogene is located on the long arm of chromosome 7 at position 34. It encodes for a serine/threonine kinase, which is involved in the RAS/RAF/MEK/ERK signaling pathway. When activated by oncogenic mutations, BRAF phosphorylates MEK and promotes cell growth, proliferation, and survival. The highest incidence of BRAF mutation is in malignant melanoma (27%–70%), followed by papillary thyroid cancer, colorectal cancer, and serous ovarian cancer. BRAF mutations have also been reported in 1% to 3% of NSCLCs. In contrast to melanoma, only half of BRAF mutations in NSCLC are V600 E mutations. Other non-V600 E mutations reported in NSCLC include G469 A (∼35%) and D594 G (∼10%). All BRAF mutations are mutually exclusive with other driver alterations, such as those of EGFR, KRAS , and ALK . BRAF -mutated NSCLC has been reported to be mostly adenocarcinoma, and in contrast to patients with EGFR mutations or ALK rearrangements who are mostly never-smokers, patients with BRAF mutations are mostly current or former smokers. Nevertheless, patients with NSCLC and BRAF V600 E mutations have a worse prognosis and lower response to platinum-based chemotherapy than patients with wild-type BRAF . These patients have benefited from treatment with BRAF and MEK inhibitors. BRAF inhibitors, such as vemurafenib and dabrafenib, have high and selective activity against the V600E-mutant BRAF kinase, with overall responses rates from 33% to 42%. BRAF and MEK inhibitors targeting BRAF mutation–positive NSCLC, such as trametinib, selumetinib, and dasatinib, among others, are currently under evaluation in clinical trials.
Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha
PI3Ks are heterodimeric lipid kinases composed of catalytic and regulatory subunits and are part of several downstream pathways involved in cell growth, transformation, adhesion, apoptosis, survival, and motility. The PIK3CA gene is located on the long arm of chromosome 3 at position 26.3. It encodes for the catalytic subunit p110 alpha of P13Ks. PKI3CA amplifications, deletions, and somatic missense mutations have been reported in many tumors including lung cancers. In fact, PIK3CA is one of the most commonly mutated oncogenes, along with KRAS , in human cancers. Mutations are found in 1% to 4% of patients with NSCLC, usually affecting exons 9 and 20 (80%). These mutations are not mutually exclusive with other driver alterations and have been reported more frequently in lung squamous cell carcinoma compared with adenocarcinoma (6.5% vs 1.5%). However, PIK3CA mutations have not shown association with any clinicopathologic features. Squamous cell carcinomas with PIK3CA gains are not accompanied by other genetic alterations, suggesting that this gene may play an important role in the pathogenesis of squamous cell cancers. Studies have shown that PIK3CA mutations in EGFR -mutated lung cancer confer resistance to EGFR-TKIs and are a negative prognostic predictor in patients with NSCLC treated with EGFR-TKIs. PI3KCA alterations and their downstream effectors, such as phosphatase and tensin homolog, mTOR, and AKT, are potential therapeutic targets for NSCLC therapy and are being evaluated in clinical trials for lung cancer. Alterations in PI3KCA are detected using sequencing approaches, mostly NGS assays.
Neurotrophic Receptor Tyrosine Kinase 1
The neurotrophic receptor tyrosine kinase 1 ( NTRK1 ) proto-oncogene is located on chromosome 1q21 to 22 and encodes for a receptor tyrosine kinase, also known as tropomyosin-related kinase (TRK) A, belonging to the TRK superfamily of receptor tyrosine kinases (117). NTRK1 is involved in the regulation of cell growth and differentiation via activation of several signal transduction pathways including MAPK, PI3K, and phospholipase C-gamma. NTRK1 rearrangements have been found in colon cancer, thyroid cancer, and glioblastoma multiforme. In lung cancer, approximately 3% of adenocarcinomas harbor NTRK1 fusions, and some fusion partners, including myosin phosphatase RHO-interacting protein ( MPRIP ) -NTRK1 and CD74-NTRK1 , have been reported. All of these fusions result in constitutive TRKA kinase activity, which has been reported to be oncogenic. In early phase 1 studies, NTRK inhibitors, such as entrectinib and LOXO-101, have shown promising results in patients with solid tumors harboring NTRK fusions.
Fibroblast Growth Factor Receptor
The fibroblast growth factor receptor ( FGFR ) gene is located on chromosome 8 at position 12 and encodes for a tyrosine kinase receptor belonging to the FGFR family. The FGFR family includes four receptor tyrosine kinases (FGFRs 1–4). When ligand-receptor binding occurs, FGFR dimerizes and phosphorylates FGFR substrate 2-alpha (FRS2α), leading to activation of different pathways, including the RAS/MAPK and PI3K/AKT/mTOR pathways, promoting cell survival, motility, invasiveness, and proliferation. In cancer, FGFR gene amplifications, somatic missense mutations, and chromosomal translocations are the most frequent mechanisms of activation. FGFR has been identified as an oncogenic driver in breast, gastric, endometrial, urothelial, and brain tumors, among others. In lung cancer, the incidence of FGFR1 amplification is significantly higher in squamous cell carcinoma (20%) compared with adenocarcinoma (3%) and is more frequent in current smokers compared with former and never-smokers. Other specific clinic-demographic features also correlate with FGFR1 amplification. Some studies have recognized FGFR amplification as an independent negative prognostic factor in patients with NSCLC, whereas other studies have shown the opposite. In addition, FGFR amplifications may be found in concurrence with other tumor genetic alterations including TP53 and PIK3CA mutation and platelet-derived growth factor receptor A ( PDGFRA ) amplification. Somatic FGFR mutations in lung tumors usually occur in FGFR2 and FGFR3 and have been detected in 6% of lung squamous cell carcinomas. Multiple FGFR inhibitors, such as ponatinib, a multitargeted kinase inhibitor that displays potent pan–anti-FGFR activity, are in development, with promising results in cell lines and xenograft models. Phase 1 and 2 clinical trials of FGFR inhibitors (eg, dovitinib, nintedanib, ponatinib, and AZD4547) are ongoing in patients with NSCLC. FGFR gene copy number is usually assayed by FISH; however, members of this family are frequently part of NGS testing panels.
Discoidin Domain Receptor Tyrosine Kinase 2
The discoidin domain receptor tyrosine kinase 2 gene ( DDR2 ) is located on the long arm of chromosome 1 at position 23.3 and encodes for a tyrosine kinase receptor that is expressed in mesenchymal tissues and that binds fibrillar collagen as ligand. DDR2 activates important signaling pathways including SRC, SRC homology domain-containing, Janus kinase, ERK1/2, and PI3K and promotes cell migration, proliferation, and survival. In cancer, DDR2 mutations have been reported in melanoma and uterine, gastric, bladder, and colorectal cancers. In lung cancer, DDR2 mutations occur in 3% to 4% of lung squamous cell carcinomas compared with 0.5% of adenocarcinomas and are only present in smokers. No other significant association with clinicopathologic status has been found. At least 11 different DDR2 mutations have been identified distributed throughout the gene and include the extracellular-binding discoidin domain and the cytoplasmic kinase domain. DDR2 mutations have been associated with response to dasatinib (a multitargeted kinase inhibitor) in preclinical models and early phase clinical trials. Phase 2 clinical trials of dasatinib in patients with lung squamous cell carcinoma are under way.
Related posts:
 Evolving Treatment Options for Lung Cancer
Evolving Treatment Options for Lung Cancer
 Second-Line Chemotherapy and Beyond for Non–Small Cell Lung Cancer
First-Line Systemic Therapy for Non–Small Cell Lung Cancer
Diagnosis and Treatment of Anaplastic Lymphoma Kinase–Positive Non–Small Cell Lung Cancer
Advances in Small Cell Lung Cancer
Systemic Treatment of Brain Metastases
Second-Line Chemotherapy and Beyond for Non–Small Cell Lung Cancer
First-Line Systemic Therapy for Non–Small Cell Lung Cancer
Diagnosis and Treatment of Anaplastic Lymphoma Kinase–Positive Non–Small Cell Lung Cancer
Advances in Small Cell Lung Cancer
Systemic Treatment of Brain Metastases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


