Class
Symbol
Characteristic
Disease/biological function associations
Long intergenic noncoding RNAs
lincRNAs
Ranging from hundreds to tens of thousands nts; lie within the genomic intervals between two genes; transcriptional cis-regulation of neighboring genes
Involved in tumorigenesis and cancer metastasis/involved in diverse biological processes such as dosage compensation and/or imprinting
Long intronic noncoding RNAs
Lie within the introns; evolutionary conserved; tissue and subcellular expression specific
Aberrantly expressed in human cancers/possible link with posttranscriptional gene silencing
Telomere-associated lncRNAs
TERRAs
100 bp–9 kb; conserved among eukaryotes; synthesized from C-rich strand; polyadenylated; form inter-molecular G-quadruplex structure with single-stranded telomeric DNA
Possible impact on telomere-associated diseases including many cancers/negative regulation of telomere length and activity through inhibition of telomerase
Long noncoding RNAs with dual functions
Both protein-coding and functionally regulatory RNA capacity
Deregulation described in breast and ovarian tumors/modulate gene expression through diverse mechanisms
Pseudogene RNAs
Gene copies that have lost the ability to code for a protein; potential to regulate their homologous protein-coding genes; made through retrotransposition; tissue specific
Often deregulated during tumorigenesis and cancer progression/regulation of tumor suppressors and oncogenes by acting as microRNA decoys
Transcribed-ultraconserved regions
T-UCRs
Longer than 200 bp; absolutely conserved between orthologous regions of human, rat, and mouse; located in especially intra- and intergenic regions
Expression is often altered in some cancers; possible involvement in tumorigenesis/antisense inhibitors for protein-coding genes or other ncRNAs
Long noncoding RNAs are a diverse group of transcripts, which differ in size, location in the genome, and other biological properties. Such diversity is also the reason why a wide range of functions was observed in lncRNAs. Long ncRNAs can affect gene expression via RNA polymerase II inhibition (B2 SINE) or chromatin modification (COLDAIR). They can also serve as precursors of siRNAs (H19) and other small ncRNAs (GAS5). By forming complexes with proteins, they may modulate its activity (SRA), influence structural and regulatory functions (XIST), change protein localization, or affect epigenetic processes (HOTAIR). They are also involved in alternative splicing of mRNA (MALAT-1) and are responsible for microRNA silencing (HULC) (See Fig. 7.1).
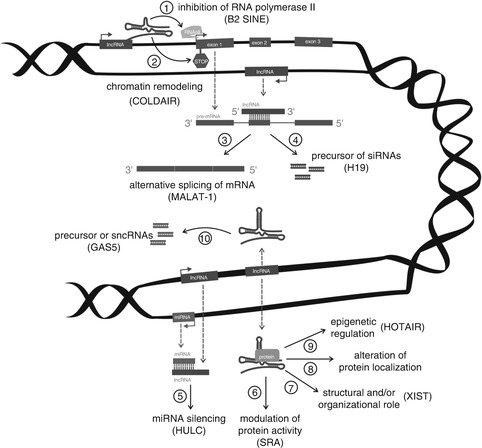
Fig. 7.1
Schematic illustration of lncRNAs functioning. lncRNA transcribed from an upstream noncoding promoter can negatively (1) or positively (2) affect expression of the downstream gene by inhibiting RNA polymerase II recruitment and/or inducing chromatin remodeling, respectively. lncRNA is able to hybridize to the pre-mRNA and block recognition of the splice sites by the spliceosome, thus resulting in an alternatively spliced transcript (3). Alternatively, hybridization of the sense and antisense transcripts can allow Dicer to generate endogenous siRNAs (4). The binding of lncRNA to the miRNA results in the miRNA function silencing (5). The complex of lncRNA and specific protein partners can modulate the activity of the protein (6), is involved in structural and organization roles of the cell (7), alters the protein localizes in the cell (8), and affects epigenetic processes (9). Finally, long ncRNAs can be processed to the small RNAs (10)
Long Intergenic Noncoding RNAs
Long intergenic noncoding RNAs (also known as lincRNAs) were originally identified by Guttman et al., who used methods to reconstruct the transcriptome of a mammalian cell [19]. They described an evolutionary conserved group of noncoding RNAs with a range in length from a few hundred to tens of thousands of bases. Genes of lincRNAs are localized in regions of DNA between two protein-coding genes, but they lack any protein-coding capacity and open reading frames. To date, more than 8,000 lincRNAs have been identified, but most of them remain unannotated [8, 20], and therefore the functions of lincRNAs are largely unknown. However, the involvement of lincRNAs in biological processes has been found, including cell-cycle regulation, imprinting, embryonic stem cell pluripotency, and cell proliferation [21, 22].
The most unclear part of lincRNAs biology is the basis of their molecular mechanism of action. In this context, targeting of chromatin modification complexes (i.e., histone-modifying enzymes) is frequently mentioned, which directly leads to gene expression regulation [8]. Some lincRNAs interact with numerous effector proteins and thus control their levels (i.e., XIST). Others can affect alternative splicing (MALAT-1) by controlling levels of splicing factors [23]. Recently, it has been demonstrated that lincRNAs can act as competitive inhibitors of microRNAs termed “microRNA sponges.” Such molecules contain binding sites for specific miRNAs and thus may regulate their level [24].
As mentioned, lincRNAs regulate significant number of genes, but their expression is under genetic control as well [25]. Individual lincRNAs are transcriptionally regulated by important transcription factors such as p53, NFκB, Sox2, Oct4, and Nanog [19]. Interestingly, Juan et al. suggest that some miRNAs may bind lincRNAs and cause their repression [26].
Relevance for translational medicine stems from the fact that long intergenic noncoding RNAs are remarkably tissue specific and deregulated within a large number of diseases, including cancer. Different levels of lincRNAs were observed at various stages of breast cancer, so based on these gene expression patterns they may serve as potential prognostic markers [9].
Long Intronic Noncoding RNAs
More than one-third of conserved noncoding regions in human genome consist of intronic regions. John Mattick first suggested that sequence of introns is not random and that introns may be involved in gene regulation [27]. According to this, it was found that about 81 % of human protein-coding genes have transcriptionally active introns [28]. Finally, discovery of numerous evolutionarily conserved regions in introns that match the size of lncRNAs led to the identification of a new class of transcripts—long intronic noncoding RNAs. These RNAs are exclusively expressed in the nucleus, and it is expected that intronic ncRNA expression would be responsive to common physiological signals, e.g., hormones [29]. The biogenesis is poorly understood, but involvement of RNA polymerase II (RNAP II) is assumed. The presence of poly(A+) tail may serve as indirect evidence [18].
The main role of intronic noncoding RNAs is posttranscriptional regulation of gene expression. In his study, Louro et al. described some mechanisms by which RNAs can regulate gene expression [30]. Interestingly, it was found that intronic ncRNAs can serve as precursors of smaller noncoding RNAs. Another mechanism is a direct interaction with promoters, which decrease the expression of the protein-coding RNA. Intronic ncRNAs can also affect RNA alternative splicing by forming RNA–RNA duplexes. Finally, they are probably able to stabilize protein-coding RNA localized on the same locus [28].
Some oncogenes or tumor suppressor genes have noncoding RNAs transcribed from their introns. This might be one of reasons why altered expression of intronic ncRNAs with various malignancies was detected. Moreover, level of long intronic ncRNAs significantly correlates with different degrees in renal, prostate, and pancreatic carcinoma [31–33].
Long ncRNAs with Dual Functions
According to the central dogma of molecular biology, RNA was considered as an intermediated molecule required for the formation of protein. After the discovery of noncoding RNAs, which in many aspects resembled the mRNA, but lack protein-coding capacity, it was obvious that RNA can act either as a functional or protein-coding molecule. Hence, it was a big surprise when bifunctional RNAs were found. Such RNAs serve both as intermediate molecules translated into protein and as functional RNA [34].
Functions of RNAs are dependent on their secondary and tertiary structure, so the presence of isoforms can be important for bifunctional character [35]. Well described is the steroid receptor RNA activator (SRA), whose RNA is a noncoding RNA that coactivates several human hormone receptors like progesterone, estrogen, and androgen. Moreover, isoforms of SRA are also expressed to produce proteins. SRA transcripts have been identified in normal human tissues, and increasingly SRA RNA is expressed in breast and ovarian tumors. Interestingly, higher levels of noncoding isoforms of SRA were observed in tumor tissue [36, 37].
Telomere-Associated lncRNAs
Telomeres—heterochromatic complexes located on linear chromosome ends—are formed by tandem repeats of the TTAGGG sequence. With each run of cell division, one telomeric hexanucleotide is lost, which finally leads to chromosome destabilization. Therefore, telomeres protect chromosomes from degradation and repair activities [38, 39]. Until recently, it was assumed that telomeres are transcriptionally silent, but a recently discovered group of long noncoding RNAs confirmed that telomeres are transcribed into telomeric repeat-containing RNA (TERRA or Tel RNA). TERRA transcripts range between 100 bp and 9 kb and originate in the subtelomeres of telomeric C-rich strand [40, 41]. Based on RNA-FISH techniques, it was identified that TERRA associates with telomeric chromatin [42]. Other studies have suggested a likely role of TERRA in the enzyme telomerase regulation [43]. Finally, TERRA seems to be involved in negative regulation of telomere length [44].
Pseudogene RNAs
For a long time, pseudogenes were considered as failed copies of coding genes that had lost the capability to produce proteins. Nevertheless, recent research has revealed the ability of pseudogenes to regulate homologous protein-coding genes [45]. Pseudogenes may arise as a result of simple mutations or be generated by retrotransposition, during which reverse-transcribed RNAs are integrated into the genomic sequence [46, 47]. Many pseudogenes are transcribed into RNA, which can be later processed into smaller RNAs. Thus, gene expression regulation is based on an RNA interference process. Interestingly, pseudogenes can affect gene expression regulation by acting as miRNA decoys [48]. Some studies provide evidence that pseudogenes (i.e., MYLKP1) are involved in cancerogenesis and suggest them as potential diagnostic and therapeutic targets in cancer [49].
Transcribed-Ultraconserved Regions
The last newly discovered class of ncRNAs is known as transcribed-ultraconserved regions (T-UCRs). Overall, 481 T-UCRs were annotated—all of them are genomic segments of more than 200 base pairs [50]. They are extremely evolutionarily conserved among mammals [51] and are localized especially in intra- and intergenic regions [52]. The degree of conservation may have a fundamental functional importance for ontogeny and phylogeny of mammals. Untranslated UCRs may serve as distal enhancers [53]; on the contrary transcripts they are involved in gene expression regulation as antisense inhibitors for protein-coding genes. Calin et al. found that the expression of many T-UCRs is altered in some types of cancer, especially in adult chronic lymphocytic leukemias, colorectal and hepatocellular carcinomas, and neuroblastomas [54]. Accordingly, it was found that T-UCRs are often located at fragile regions of chromosomes. Specific transcribed-ultraconserved regions are also associated with prognosis and response to therapy, which makes them promising targets in cancer research [53, 55].
Methods for High-Throughput Analysis of Long Noncoding RNAs in Cancer
The human genome mapping has revealed that more than 90 % of the genome is transcribed. Application of high-throughput techniques highlighted the complexity of mammalian transcriptome and led to the discovery of long noncoding RNAs, a class of regulatory noncoding RNAs [16, 56]. As a transcriptional class, lncRNAs were first described during the large-scale sequencing of full-length cDNA libraries in the mouse [13]. Such large-scale cDNA analysis and genome annotations can detect or predict thousands of lncRNAs, but their biological functions remain, in most cases, unknown [57]. Methods used in the study of lncRNAs can be thus divided by purpose into (1) high-throughput methods designed for lncRNAs identification (microarrays, RNA sequencing), (2) methods designed for verification of high-throughput data (qRT-PCR, northern blot, FISH, RNAi), and (3) methods designed for detection of RNA–protein interactions (RIP, RIP-CHIP); see Fig. 7.2 [58].
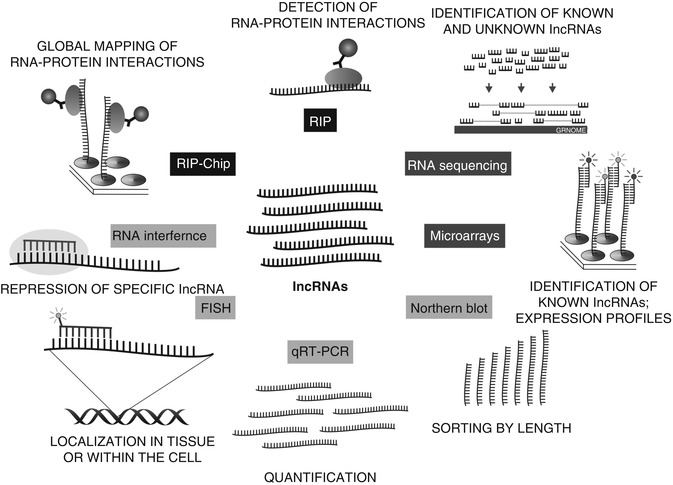
Fig. 7.2
Methods used in the study of lncRNAs can be divided into (1) high-throughput methods designed for lncRNAs identification (microarrays, RNA sequencing), (2) methods designed for verification of high-throughput data (qRT-PCR, northern blot, FISH, RNAi), (3) methods designed for detection of RNA–protein interactions (RIP, RIP-CHIP)
Microarrays
The structure and expression of long noncoding RNAs is very similar to mRNAs, even though lncRNAs lack open reading frames and other properties necessary for them to be translated into proteins [59]. One of the most common methods of identification is the microarray-based approach. Microarrays are based on nucleic acid hybridization between target molecules and probes and enable simultaneous monitoring of thousands of genes in a single experiment. However, this method shows only whether or not lncRNA is expressed and is therefore not suitable for the identification of novel transcripts [60]. On the other hand, microarrays allow us to detect differences in transcriptional profiles between different tissues and cell types or identify possible targets of lncRNAs [21, 58].
RNA Sequencing
The transcriptome includes all RNAs synthesized in an organism, including protein-coding, non-coding, alternatively spliced, alternatively polyadenylated, alternatively initiated, sense, antisense, and RNA-edited transcripts [13]. The most widely used method for qualitatively and quantitatively profiling the full set of transcripts is RNA sequencing (RNA-seq), which is based on next-generation sequencing (NGS) [61]. RNA-seq works on a genome-wide scale at single nucleotide resolution and is not limited to detecting already known sequences. Thus, it can be used to discover previously unknown lncRNAs [62]. Considerable disadvantages of this approach are the time and cost related to the downstream analysis of the RNA-seq data [60]. After sequencing, the generated reads are used to assemble the transcriptome, and then novel lncRNAs can be identified and annotated via bioinformatic databases (i.e., FANTOM or ENCODE) [63]. After that, novel lncRNAs often undergo further scrutiny to verify that they are not transcriptional noise and that they indeed do not encode proteins. In the same way, candidate targets are required to be verified by other molecular biology methods as well.
qRT-PCR and Northern Blot
Quantitative real-time polymerase chain reaction (qRT-PCR) allows amplification and quantification of selected segments of the genome. This highly sensitive method is used for gene expression studies, but qPCR can be used even for analysis of lncRNAs. For this purpose, qPCR is combined with reverse transcription (RT), which ensures transcription of RNA into cDNA. For the verification of high-throughput data, qRT-PCR is frequently followed by northern blot analysis, which is the only direct method to prove the presence of RNA without the need of amplification. This combination was used to demonstrate the lengths of detected lncRNAs and their level of expression [64].
Fluorescence In Situ Hybridization
Fluorescence in situ hybridization (FISH) is a type of hybridization that uses a fluorescently labeled complementary DNA or RNA strand to localize a specific sequence on a chromosome, section of tissue (in situ) fixed on a slide or even cell [65]. In lncRNAs research, FISH was used to detect individual lncRNA or their localization within the cell [23, 66].
RNA Interference (RNAi)
The process of RNA interference involves the binding of short interfering RNA molecules to mRNAs, which leads to expression–repression of a gene of interest. The use of synthetic dsRNA allows also effective knockdown of specific lncRNAs and is very important for studying their functions [67].
RNA Immunoprecipitation and RIP-Chip
Long noncoding RNA may affect the regulation of gene expression also through the modification of chromatin via interactions with various proteins (i.e., transcription factors). Such RNA–protein interactions are easy to detect with RNA immunoprecipitation (RIP). It is an antibody-based technique in which the RNA-binding protein of interest immunoprecipitates together with its associated RNA and allows localize RNA-binding sites on the genome [68]. A number of lncRNAs, such as Xist and Tsix, were identified through this approach [69].
Major advances in the global analysis of subsets of mRNAs bound to RNA-binding proteins brought combinations of RIP and microarrays called RNA-binding protein immunoprecipitation-microarray (Chip) profiling or RIP-Chip [70]. Using this high-throughput method, it was revealed that a large number of lincRNAs associate with chromatin-modifying complexes to affect gene expression [8].
Long Noncoding RNAs in Pathogenesis of Breast Cancer
lncRNAs were found to be deregulated in several human cancers and show analogically to protein-coding genes tissue-specific expression. Functional studies elucidated a large range of molecular mechanisms used by lncRNAs in cancer cells. Till now, only a few lncRNAs were observed to have altered expression in breast cancer, including HOTAIR, MALAT-1, GAS5, ZFAS1, LSINCT5, SRA1, H19, XIST, and BC200, which are characterized in detail in Table 7.2. Here we discuss molecular functioning of these lncRNAs mainly in the context of typical hallmarks of cancer. Some important molecular mechanisms used by particular lncRNA are mentioned although they are observed in cancer types other than breast cancer.
Table 7.2
Characteristics of lncRNAs deregulated in breast cancer
Gene | Locus | Length (kb) | Function in cancer | Association in hallmarks of cancer | Tumor types | References |
|---|---|---|---|---|---|---|
HOTAIR | 12q13 | 2.2 | Oncogene | Activating invasion and metastasis | BC, CRC, PaC, HCC | [73] |
MALAT-1 | 11q13 | 8.4 | Oncogene | Activating invasion and metastasis; evading growth suppressors | BC, BlaC, HCC | |
GAS5 | 1q25 | 5.3 | Tumor suppressor | Evading growth suppressors | BC, M, Ly, PC | |
ZFAS1 | 20q13 | 0.5 | Tumor suppressor | Evading growth suppressors | BC | [84] |
LSINCT5 | 5p15 | 2.6 | Oncogene | Sustaining proliferative signaling | BC, OC | [85] |
SRA1 | 5q31 | 0.87 | Oncogene | Sustaining proliferative signaling | BC, OC, PC | |
XIST | Xq13 | 42 | Oncogene | Activating invasion and metastasis | BC, Ly, OC, CRC | |
H19 | 11p15 | 3.5 | Oncogene, tumor suppressor | Sustaining proliferative signaling | BC, BlaC, CerC, CRC, HCC, LC, M | |
BC200 | 2p21 | 0.26 | Oncogene | Activating invasion and metastasis | BC | [105] |
HOTAIR
HOX transcript antisense intergenic RNA (HOTAIR) has very important role in cancer metastasis. It was discovered as a 2.2 kb-long ncRNA transcribed in antisense direction from the HOXC gene cluster [71]. HOTAIR functions in trans by interacting and recruiting the polycomb repressive complex 2 (PRC2) to the HOXD locus, which leads to transcriptional silencing across 40 kb. PRC2 complex consisted of H3K27 methylase EZH2, SUZ12, and EED (see Fig. 7.3) [72]. Polycomb group proteins are involved in repression of transcription of large groups of genes. This pathway influences differentiation, pluripotency, and cancer development [73]. Later it was found that HOTAIR interacts with a second histone modification complex, the LSD1/CoREST/REST complex, which coordinates targeting of PRC2 and LSD1 to chromatin for coupled histone H3K27 methylation and K4 demethylation [72]. Overexpression of HOTAIR in epithelial cancer cells alters H3K27 methylation via PRC2 and therefore alters target gene expression. This leads to increased cancer invasiveness and metastases. Therefore, HOTAIR depletion inhibits breast cancer invasiveness [73].
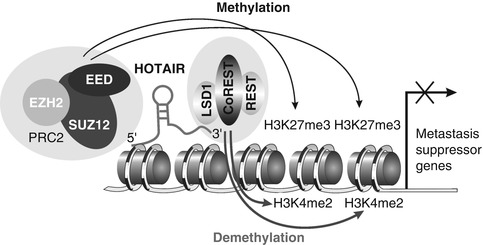
Fig. 7.3
Association of HOTAIR with the polycomb repressive complex 2 (PRC2) and LSD1/CoREST/REST complex
MALAT-1
Metastatic-associated lung adenoma transcript 1 (MALAT-1) is abundant in many human cell types. It is probably a very important transcript because its sequence is very conserved across many species. MALAT-1 is an 8,708 nt-long transcript occurring in the nucleus and frequently localized in nucleus speckles [74]. These structures play a role in pre-mRNA processing. Recently MALAT-1 has been shown to regulate alternative splicing of pre-mRNA by modulating the levels of splicing factors. These factors regulate tissue-specific alternative interactions with SR splicing factor, SRSF1, which affects the distribution of these and other splicing factors in nuclear speckle domains. Depletion of MALAT-1 with antisense oligonucleotides or transient overexpression of SRSF1 changes the alternative splicing of the endogenous pre-mRNAs. Importantly, MALAT-1 controls cellular phosphorylation status of SR proteins, thereby regulating cellular ratio of phosphorylated versus dephosphorylated form of SR proteins [23], suggesting that MALAT-1 regulates pre-mRNA processing by modulating the levels of active SR proteins. Depletion of MALAT-1 alters the processing of a subset of pre-mRNAs, which play important roles in cancer biology [75].
Recent studies indicate additional functions for MALAT-1 in the nucleus. MALAT-1 was shown to interact with the unmethylated form of CBX4, which controls relocation of growth-control genes between polycomb bodies and interchromatin granules, places of silent or active gene expression, respectively. Altered expression levels of MALAT1 were detected in breast cancer tissue compared to normal breast tissue. Also MALAT-1 locus is frequently altered in breast cancer and other tumor types [76].
GAS5
Growth arrest-specific 5 (GAS5) is the host gene for many snoRNAs, which were found in GAS5 introns, dedicating GAS5 to be involved in the important cellular activities [77]. It was proved that GAS5 transcripts displayed many different patterns of alternate splicing, but there is no putative open reading frame [78]. GAS5 functions as “riborepressor” of the glucocorticoid receptor (GR), influencing cell survival and metabolic activities during starvation by modulating the transcriptional activity of the GR. Its transcript interacts with the DNA-binding domain of GRs and reduces the probability of steroids’ interaction with their receptors [79]. In this way, GAS5 suppresses expression of several genes including cellular inhibitor of apoptosis 2 (cIAP2) and thus sensitizes cells to apoptosis. This induction of apoptosis is independent of other stimuli in several breast cancer cell lines [80]. It was also shown that silencing of endogenous GAS5 levels in breast cancer cells leads to resistance to apoptosis and various GAS5 transcripts stimulate apoptosis through different cellular signaling pathways [81]. Moreover, in leukemia cell models, GAS5 is required for normal functioning of mammalian target of rapamycin (mTOR) pathway that controls cell growth and proliferation also in breast cancer [82]. In addition, GAS5 locus was found to be frequently altered in many types of cancer (e.g., melanoma, lymphoma, prostate cancer) [83].
Zfas1/ZFAS1
Zinc finger antisense 1 (Zfas1) is a mouse antisense RNA to NFX-1 type containing zinc finger. Zfas1 is located close to a protein-coding gene and in its introns hosts three small nucleolar RNA (snoRNA) genes: Snord12, Snord12b, and Snord12c [84]. Knockdown of Zfas1 in a mammary epithelial cell line resulted in increased cellular proliferation and differentiation. But this knockdown did not substantially alter the levels of the SNORDs. Functional role for Zfas1 in the regulation of alveolar development and epithelial cell differentiation in the mammary gland, together with its dysregulation in human breast cancer, suggests ZFAS1 as a putative tumor suppressor gene. ZFAS1 is highly expressed in the mammary gland and is downregulated in breast tumors compared to normal tissue. While there is relatively low level of primary sequence conservation between Zfas1 and its human ortholog ZFAS1, secondary structures of Zfas1 and ZFAS1 transcripts share several similar features. It was proved that from ZFAS1 mRNA originates at least five different isoforms through alternative splicing.
LSINCT5
Long stress-induced noncoding transcript 5 (LSINCT5) is greatly overexpressed in many of the breast cancer cell lines [85]. LSINCT5 is polyadenylated RNA that is transcribed from the negative strand by RNA polymerase III with no open reading frame. When nuclear and cytoplasmic LSINCT5 levels are compared, LSINCT5 indicates higher expression in the nuclear fraction. Instead of decrease in cellular proliferation, knockdown of LSINCT5 in cancer-derived cell lines causes expression deregulation of several genes including important kinase (PDPK1), nuclear assembly genes (NEAT1 and PSPC1), genes involved in membrane transport (HERC1), transcription factor (ANKF41), and genes associated with carcinogenesis (EPPK1), cellular stress (PRKAA1/AMPK), motility (ACTR2), and T-cell differentiation (CXCR4, MAPK9/JNK2) [86]. Moreover, LSINCT5 is overexpressed in breast and ovarian cancer tissue.
SRA1
Steroid receptor RNA activator (SRA) modulates activity of steroid receptors and other transcription factors both at the RNA (SRA) and the protein (SRAP) level [87]. SRA appears highly expressed in the liver, skeletal muscle, adrenal gland, and the pituitary gland, whereas intermediate expression levels are seen in the placenta, lung, kidney, and pancreas. Interestingly, brain and other typical steroid-responsive tissues such as the prostate, breast, uterus, and ovary contained low levels of SRA RNA [88]. SRA is a component of ribonucleoprotein complexes recruited to the promoter of regulated genes. These complexes may contain positive regulators, such as the steroid receptor coactivator 1 (SRC-1), the DExD/H box family of RNA-helicase members p68 and p72, or the pseudouridine synthases Pus1p and Pus3p. Negative regulators, such as SMRT/HDAC1-associated repressor protein (SHARP) and the recently identified SRA stem-loop interacting RNA-binding protein (SLIRP), can also interact with SRA to decrease its activity [89]. Elevated levels of SRA are found in breast tumors and the increased SRA levels might contribute to the altered ER/PR action that occurs during breast tumorigenesis. The SRA1 gene might not only act as an ncRNA but also codes a protein that acts as a coactivator or corepressor. The ratio between noncoding and coding transcripts of SRA1 characterizes specific tumor phenotypes but might also be involved in breast tumorigenesis and tumor progression by regulating the expression of specific sets of genes [90]. The sequence of the protein encoded by SRA, referred to as SRAP, is highly conserved in Chordata. The most conserved amino acids define two distinct domains (N- and C-terminal) that represent the typical signature of this new family of proteins, and which are likely both participating in SRAP function [91]. This protein is also ubiquitously found in human cancer cell lines derived from the breast [92], the prostate [93], and other tissues, even though levels of expression appear to vary from one cell type to another.
Related posts:
 Gynecologic Considerations for Women with Breast Cancer
In Silico Disease Models of Breast Cancer
Molecular Diagnosis of Metastasizing Breast Cancer Based Upon Liquid Biopsy
Gynecologic Considerations for Women with Breast Cancer
In Silico Disease Models of Breast Cancer
Molecular Diagnosis of Metastasizing Breast Cancer Based Upon Liquid Biopsy
 Breast Circulating Tumor Cells: Potential Biomarkers for Breast Cancer Diagnosis and Prognosis Evaluation
Breast Circulating Tumor Cells: Potential Biomarkers for Breast Cancer Diagnosis and Prognosis Evaluation
 Breast Cancer Gene Therapy
Breast Cancer Gene Therapy
 Exhaled Volatile Organic Compounds as Noninvasive Markers in Breast Cancer
Exhaled Volatile Organic Compounds as Noninvasive Markers in Breast Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


