FIGURE 1 | The INK4-ARF signaling network. Physiologic mitogenic signals (green light, top left) stimulate the transcription of genes encoding D-type cyclins and facilitate their assembly into stable complexes with cyclin-dependent kinases (CDK4 and CDK6). These kinases promote the initial phosphorylation of RB and other RB family members (p130 and p107), canceling their negative regulation of E2F transcription factors and triggering an E2F-dependent program that stimulates entry into the DNA synthetic (S) phase of the cell division cycle. E2F-responsive genes include those encoding cyclins E and A, which assemble with CDK2 to enforce RB-family protein phosphorylation and drive S-phase entry. Aberrant thresholds of hyperproliferative signals emanating from constitutively active oncogenes (magnified red light, bottom) activate INK4-ARF gene expression to inhibit the activities of cyclin-dependent kinases and HDM2. ARF-mediated inhibition of HDM2 E3 ubiquitin ligase activates the p53 transcriptional program, leading either to apoptosis or cell cycle arrest. Apart from the INK4 proteins, another key mediator of cell cycle arrest is the p53-responsive CDK2 inhibitor, p21Cip1. Multiple types of DNA damage activate p53, including DNA replication errors triggered by oncogenes (bottom right). Many feedback loops regulate the network. Inactivation of p53 leads to increased ARF expression; loss of RB leads to increased p16INK4A levels (not shown). At least one of the transcription factors activating the ARF gene is E2F.
Unlike RB which is largely controlled by physiologic cues, p53 is induced by stress, typically involving genomic damage incurred in response to DNA replication errors, irradiation and genotoxic drugs, failure of the mitotic spindle checkpoint, telomere attrition, hypoxia, reactive oxygen species, and oncogene activation. Genes induced by p53 include those encoding CDK inhibitors (CDKN1A, p21Cip1), proapoptotic proteins, and MDM2 which inhibits p53-induced transcription and ubiquitinates p53 to target its degradation and terminate the p53 response. Activation of p53 initiates a program of gene expression that leads to cell cycle arrest or apoptosis, thereby eliminating incipient cancer cells.3,4
Two genes within the INK4-ARF locus—INK4A and INK4B—encode polypeptide inhibitors (p16INK4A and p15INK4B) of CDK4 and CDK6, preventing the initial G1-phase phosphorylation of RB family proteins to maintain them in their growth-suppressive state. The third gene (ARF) specifies a distinct protein (p14ARF in humans and p19Arf in the mouse) encoded in part by an alternative reading frame within the second of three exons that comprise the INK4A gene (Figure 2). The ARF protein antagonizes the E3 ligase activity of MDM2 to activate the p53-mediated transcriptional program. Because the INK4A and INK4B genes were defined before ARF was discovered, they were named CDKN (CDK inhibitor)-2A and –2B that, in retrospect, designate only the RB-dependent activities of two of three genes in the cluster. Given the role of INK4-ARF in modulating activities of RB and p53, it is not surprising that deletion of the locus is frequently detected in many distinct tumor types. But, given its compact size and apparent ease of inactivation, why has this TSG cluster been evolutionarily conserved in mammals?
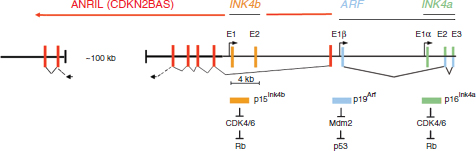
FIGURE 2 | Expanded view of the INK4-ARF locus. The two INK4 genes and ARF are schematically drawn to scale. Rectangles indicate coding exons of the three genes separated by intronic sequences (black horizontal line). Exons 2 (E2) and three (E3) of the INK4A gene (far right) are translated in alternative reading frames to generate the p16Ink4a protein (green exons and bar) and p19Arf protein (blue exons and bar, p14ARF in human cells). Promoters 5′ to INK4b exon 1 (E1) and to the alternative ARF and INK4a 5′ exons (E1β and E1α, respectively) are noted by arrows. The INK4 proteins are shown to inhibit CDK4/6 to maintain Rb in its growth-suppressive mode. By inhibiting Mdm2, p19Arf activates p53. A long intragenic noncoding RNA (designated ANRIL or CDKN2BAS) is transcribed from the ARF promoter (or from an unidentified promoter element close to exon E1β) in an antisense direction with respect to the primary INK4 and ARF transcripts. ANRIL transcripts are spliced, and putative exons are indicated by red rectangles. A ~60 kb segment within the 100 kb gap region illustrated in the schematic includes single nucleotide polymorphisms on human chromosome 9p21 in proximity to CDKN2A/B that are associated with susceptibility to coronary artery disease, aortic aneurysm, stroke, and type II diabetes.
INK4-ARF-MEDIATED TUMOR SUPPRESSION
Barriers to Cellular Self-renewal
Unlike immortal cancer cells, cultured primary cells exhibit a limited proliferative capacity and eventually undergo replicative senescence. Early investigations of DNA tumor viruses led to the realization that coinactivation of RB and p53 greatly extend cellular life span.5 The T antigen of SV40, the E6 and E7 oncoproteins of human papilloma viruses, and the E1A and E1B oncoproteins of adenoviruses disrupt both RB and p53 function, preventing exit from the cell cycle and allowing further population doublings. Notably, many primary rodent somatic cells can bypass senescence and be established as continuously growing cell lines either following DNA tumor virus infection or coinactivation of RB and p53, or Ink4-Arf deletion. In contrast, human fibroblasts fail to generate established cell lines in response to inactivation of the RB-p53 signaling network.
Although RB-p53 coinactivation extends the life span of cultured primary human cells, they eventually enter a second phase of ‘crisis’ from which only few immortalized clones emerge.6 These species-specific differences between rodent and human cells depend upon the respective lengths of chromosomal telomeres which are much longer in laboratory mouse strains than in human cells, and which, when critically shortened after repeated cell divisions, trigger chromosomal end-to-end joining, fusion-bridge-breakage cycles, mitotic catastrophe, and cell death.6–8 Continuously proliferating human cells are more precipitously called upon to solve ‘the telomere end-replication problem’, usually by reactivating telomerase or using alternative recombinational mechanisms to protect the ends of chromosomes.6 Inactivating telomerase in the germline of laboratory mice leads to symptoms of DNA damage and organismal aging only after the deficient strains are interbred for multiple sequential generations, which eventually shortens their telomeres and ‘humanizes’ them.7,8 Strikingly, these deficiencies in late-generation telomerase-deficient mice are reversed by reinstating telomerase activity.9 Thus, unlike more permissive rodent cells, the immortalization of human cell strains following inactivation of RB-p53 or INK4-ARF is highly subject to the additional restraints imposed by telomere attrition.
TSG Activities of INK4 and ARF Genes in Humans and Mice
Deletion of INK4-ARF in cancers provoked attempts to ascertain which product(s) of the locus limit the formation or progression of different tumor types. INK4A undergoes inactivating point mutations in various human cancers,10–12 and mutations that target exon1α (Figure 2) leave ARF coding sequences unaffected, underscoring the primacy of INK4A in these tumors. In mice, the germline deletion of Ink4a alone predisposes animals to spontaneous tumorigenesis and accelerates the appearance of carcinogen-induced cancers.13 Few spontaneous tumors arise in a strain carrying an Ink4a point mutation that is frequently detected in human tumors, although interbreeding these animals to Arf heterozygotes sensitizes them to carcinogen-induced melanomagenesis.14 Mutations that disable p16INK4A perturb the structure of tandem ankyrin repeats that make up the body of the protein, resulting in its inability to bind and inhibit CDK4 and CDK6.15 For reasons that remain unclear, mutations targeting INK4B in human tumors are far less common.10–12 In mice, disruption of Ink4b alone has only modest phenotypic effects, but Ink4b compensates for Ink4a inactivation and plays a more crucial role when p16Ink4a is absent.16 The presence of two other chromosomally unlinked INK4 family members, CDKN2C (INK4C) and CDKN2D (INK4D), that encode proteins with CDK inhibitory activities biochemically indistinguishable from those of INK4A and INK4B, further complicate the picture.10 INK4C has TSG activity and can compensate for INK4A loss in some cancers,17 but INK4D lacks documented TSG activity, and its disruption in mice is well tolerated.
The ARF protein bears no structural relationship to INK4 proteins. It is unusually basic and composed of approximately 20% arginine residues; the polypeptide exhibits no recognizable motifs, is natively unstructured, and acquires activity and stability only when bound to targets, such as MDM2.18 ARF can bind to many proteins, but the biological significance of most such interactions remains controversial. At least one other associating protein, nucleophosmin (NPM/B23), binds p19Arf with high stoichiometry, targets it to nucleoli, and stabilizes p19Arf by preventing its N-terminal ubiquitination and proteasomal degradation. NPM-Arf complexes do not contain Mdm2, and vice versa. Although most tumor suppressor activity of p19Arf is mediated through the Mdm2-p53 axis, p19Arf also has p53-independent activities, including an ability to induce sumoylation of other proteins, Mdm2 and NPM among them.18 The N-terminal domain of p19Arf, encoded in toto by exon-1β and comprising only a little more than 60 amino acid residues, is sufficient to bind to and inhibit Mdm2 E3 ligase activity, thereby inducing p53. Together, these features suggest that missense mutations that disable ARF function are improbable and, indeed, its specific inactivation by mutation is relatively rare.11
In murine cancers, targeted deletion of Arf alone (with retention of Ink4 gene function) is highly oncogenic19 and results in comparatively greater spontaneous tumor penetrance than inactivation of Ink4a or Ink4b.13,14,16 Yet, all three genes manifest TSG activities, and their codeletion leads to more dramatic effects than the inactivation of any one alone.16,20 In humans, the fact that INK4A is frequently mutated, whereas ARF alone is not, has led to the general impression that alterations affecting p16INK4A play the prominent role in tumor suppression, whereas Arf figures more importantly in murine tumors. Nonetheless, given that frequent INK4–ARF deletions and p53 mutations occur as mutually exclusive events in certain human cancers, a parsimonious interpretation is that ARF interacts epistatically with p53 to suppress evolution of these tumor types.
Although the INK4 and ARF proteins separately target the RB and p53 ‘pathways’ (Figure 2), there is significant cross talk within the signaling network (Figure 1). For example, dismantling RB, either through INK4 mutation or cyclin D-CDK overexpression, or RB loss per se results in robust E2F activation of the ARF promoter,21 invoking ARF-MDM2-p53 signaling to protect cells from RB loss of function. In turn, p53-mediated induction of p21Cip1, a potent inhibitor of CDK2, leads to reduced RB phosphorylation, promoting cell cycle arrest. The fact that p16INK4A levels rise in response to RB inactivation, and that ARF expression is greatly increased when p53 function is abrogated, provides further examples of feedback control.22 Thus, the signaling circuits modulated by INK4-ARF are highly interconnected.
The configuration of the INK4-ARF genes is conserved in mammals, including the alternative exon-2 codon usage of INK4A and ARF. However, these features are not conserved in lower vertebrates.12 For example, only a rudimentary ARF gene encoded almost in its entirety by exon-1β is found in chickens, which lack exon-1α, do not encode p16INK4a, but retain an INK4B ortholog. In contrast, an INK4B-like gene is found in fish, but ARF is absent. On the basis of nucleotide sequence and species comparisons, it has been suggested that INK4A arose by duplication of INK4B and that ARF exon-1β was acquired subsequently. In mammals, coordinate epigenetic regulation of the three closely spaced promoters (Figure 2) may confer an evolutionarily acquired advantage (see below).12
Stress-induced Activation and Mechanisms of INK4-ARF Tumor Suppression
Expression of INK4-ARF is not detected in most somatic tissues of young mammals, but it can be induced by various forms of oncogenic stress. Observations that an activated RAS oncoprotein induced both p16INK4A and p53 in primary fibroblasts provided the first evidence linking oncogenic signaling with INK4A induction.23 Expression of INK4A in response to constitutively active mutant forms of RAS has been well documented in cultured human and mouse primary cells, as well as in precancerous lesions from patients. In human cells, enforced RAS–RAF pathway activation has been reported to induce p16INK4A, but not p14ARF, whereas Ink4a and Arf tend to be coregulated by mutant Ras in rodents.11,12
While remaining unresponsive to physiologic levels of mitogenic signaling, the entire murine Ink4–Arf locus is poised to respond to aberrant thresholds of hyperproliferative signals emanating from constitutively activated oncoproteins1,4 (Figure 1). Notably, oncogene-induced triggering of Ink4-Arf gene expression is an indolent process. This suggests that the locus is insulated in responding to acute stress signals but undergoes chromatin remodeling and is progressively induced in the course of chronic oncogene stimulation. In mice, the general paradigm is that Arf loss of function, even when Ink4 genes are conserved, collaborates with oncogene activation to accelerate tumor progression and metastasis, closely mimicking effects of p53 inactivation. Functional collaboration between Arf inactivation and different upregulated oncoproteins, including Ras, Myc, mutant EGF and PDGF receptors, Bcr-Abl, Wnt, and others in mouse models, strongly supports the view that the Arf-Mdm2-p53 axis monitors mitogenic signal intensities and that Arf loss, in turn, relieves oncogene-initiated cancer cells from such restraint.1,4
One outcome of oncogene-induced p53 and p16INK4A activation is the induction of cellular senescence in vivo leading to durable cell cycle arrest and the eventual elimination of senescent cells.23 Senescence is characterized by lack of responsiveness to mitogenic growth factors, extensive chromatin remodeling (appearance of senescence-activated heterochromatic foci), and global gene expression changes associated with characteristic alterations in cellular morphology, production of particular inflammatory cytokines, and upregulation of biomarkers, such as senescence-activated β-galactosidase.24,25 These features distinguish senescent cells from quiescent (G0) populations that can reenter the cell division cycle when stimulated with appropriate mitogens. Moreover, establishment of the senescent state seems to be a relatively slow process that requires the prolonged maintenance of p16INK4A-induced inhibition. Senescence provides a barrier to tumor formation and has been documented in premalignant tissues in rodents and humans.25–27 The host immune system is activated by cytokines released from senescent cells, triggering their elimination.25,27 In mice, both Ink4a and Arf can elicit oncogene-induced senescence; however, p16INK4A appears to play the central role in human cells.11,26
Apart from impinging on the INK4-ARF locus, oncogene activation in early premalignant tumors increases the frequency of DNA replication errors, resulting in a DNA damage response that activates p5328,29 (Figure 1). Analysis of human precancerous lesions has indicated that markers of senescence and DNA damage are frequently concordant, implying that (ARF-independent) ATM/ATR kinase signaling strongly contributes to oncoprotein-mediated p53 activation. A frequently misunderstood distinction is that while p53 is activated by acute DNA damage, Arf is not.18,19 In turn, p53 activity is readily induced by DNA damage in Arf-null cells.19 In marked contrast, many chronic DNA damage signals have been reported to induce p16INK4A,11 further implying that various signaling inputs differentially engage INK4A and ARF.
Given that DNA damage stems from both oncogene-induced senescence,25–29 and telomere dysfunction,30,31 each of which has been associated with organismal aging,11,31,32 Arf would seem unlikely to be directly involved in these particular processes. Reinforcing this view, responses to telomere shortening, DNA damage, and degenerative aging that are reversed by p53 deletion in mice33 are not attenuated by disruption of Ink4a-Arf.34 If, in fact, DNA damage is central to malignant transformation, the p53-dependent (Arf-independent) DNA damage response should bear major responsibility for tumor suppression. This has been challenged by experiments in which a p53 allele that could be toggled between inactive and active states was functionally restored at different times after mice were exposed to ionizing radiation. In this setting, widespread p53-dependent apoptosis induced by DNA damage did not contribute to tumor suppression, whereas restoration of p53 after the DNA damage response had subsided was potently tumor suppressive and depended upon p19Arf.4
Key questions therefore concern the degree to which p53 is activated by oncogene-induced DNA damage, by ARF, or through both pathways in precancerous tissues, and whether there are significant species-specific differences in the manner by which the RB/p53/INK4-ARF tumor suppressor network responds to stress signals in humans and mice. Whatever the circuitry, oncogenic stress not only provides the force that induces INK4-ARF gene expression but also selects for the emergence of rare cells that delete the locus and subsequently escape tumor suppression. Thus, the finding that a cancer cell has sustained an INK4-ARF deletion may be taken as evidence that the locus was activated at some earlier stage of tumor development.
INK4-ARF AND STEM CELL SELF-RENEWAL
Silencing of INK4-ARF in Stem Cells
Self-renewal of dividing stem cells requires mechanisms that maintain both their pluripotent state and capacity to differentiate. In embryonic stem (ES) cells, pluripotency is promoted by an autoregulatory circuit of ‘core factors’ (principally Oct4, Sox2, and Nanog) that repress cell differentiation.35 In turn, enforced expression of Oct4 and various combinations of other transcription factors, including Sox2 and Nanog, can reprogram somatic cells into induced pluripotent stem (iPS) cells, reversing the epigenetic landscape and resetting it to an ES-like state. ES cell division cycles are characterized by a short G1 phase marked by little or no hypophosphorylated RB, desensitizing them to CDK regulation by mitogens or, conversely to Ink4 inhibitors. ES cells also resist p53-dependent cell cycle arrest. Hence, they are relatively refractory to Ink4-Arf-mediated TSG activity (Box 1).
INK4-ARF REGULATION AND STAGE-SPECIFIC TUMOR SUPPRESSION
Cardinal features of cell cycle control and regulation of the Ink4-Arf/RB/p53 network are indicated. HSCs and pro-B cells exemplify several well-defined differences between tissue stem cells and their more differentiated progeny.
Embryonic stem cells
- G1 interval is short.
- D-type cyclin levels are low.
- RB is constitutively hyperphosphorylated by cyclin E/A-CDK2.
- p53 is insulated from activation by DNA damage.
- Ink4-Arf locus is silenced.
Hematopoietic stem cells (fetal and young adult)
- G1 interval is regulated by environmental cues.
- D-type cyclins are essential.
- RB is sequentially phosphorylated by cyclin D/E/A-CDKs.
- p53 is acutely activated by DNA damage (Atm/Atr pathways).
- Ink4-Arf locus is silenced.
Pro-B cells (young adult)
- G1 interval is regulated by environmental cues.
- D-type cyclins are essential.
- RB is sequentially phosphorylated by cyclin D/E/A-CDKs.
- p53 is acutely activated by DNA damage (Atm/Atr pathways).
- Ink4-Arf locus responds to hyperproliferative signals (Ink4a, Arf) and DNA damage (Ink4a); deletion promotes cancer.
HSCs and pro-B cells (aged adult)
- Stress-induced DNA damage and cumulative mutations trigger apoptosis and cellular senescence.
- Ink4-Arf expression increases with age.
- Tissue regenerative capacity is diminished.
- Cells resist oncogene-induced transformation, but disruption of Ink4-Arf/RB/p53 network predisposes to cancer.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


