Fig. 28.1
The central tenants of induction of an in situ immune response to tumor antigen. First is a stimulus causing immunogenic cell death, which leads for resident or recruited APCs to take up antigen and become activated due to pro-inflammatory signals, which are both an innate immune response and antigen processing and presentation/cross-presentation to achieve an adaptive immune response
28.2 Immunogenic Cell Death
Immunogenic cell death results in release of tumor antigens from dying cells into the microenvironment along with cell-stress signals and damage-associated molecular patterns (DAMPs) capable of activating receptors on and in APCs. As opposed to apoptosis, a comparatively tolerogenic form of cell death, immunogenic cell death, not only provides the antigen but also provides inflammatory stimuli, and as such, agents inducing this form of toxicity are occasionally able to elicit a vaccinal response on their own [15].
While immunotherapy is generally described as mechanistically distinct, in reality there may have been an immunostimulatory component to many of the cytotoxic therapies that have been employed over the last century. An early observation of the role of the immune system in cancer was the abscopal effect, in which localized radiation given to a patient resulted in distant regression of disease outside of the radiation field [16]. While rare, this abscopal effect offers a proof of principle that can be further optimized. Radiation has been used in a variety of vaccine/immunotherapy regimens given its ability to induce immunogenic cell death. Radiation stimulates translocation of calreticulin to the cell surface and release of a multitude of DAMPs, including free DNA, high-motility group box 1 (HMBG1), and ATP; these signals stimulate pro-inflammatory receptors on dendritic cells (DCs), activating the inflammasome and Toll-like receptor (TLR) pathways among other pattern recognition receptors (PRRs) [17–21]. Upon uptake of dead or dying tumor cells by phagocytes, cell-free DNA is also capable of activating the stimulator of interferon (IFN) gene (STING) pathway, inducing IFN production and potentiating activation of a strong effector T-cell priming [22]. In parallel, radiation further induces expression of key chemokines that are critical for recruitment of effector CD8+ T cells to the irradiated tumor, including CXCL9, CXCL10, and CXCL16 [23, 24]. At the same time, radiation has also been shown to suppress the immune response through recruitment and differentiation of regulatory T cells and myeloid-derived suppressor cells (MDSCs) to the tumor microenvironment, leading to secretion of cytokines such as TGF-β and conversion of immunogenic ATP into adenosine by CD39 and CD73, which suppresses T effector cell activation and survival via the A2a adenosine receptor [25–27].
Our group and others have demonstrated a significant upregulation of PD-1 and PD-L1 on intratumoral T cells and tumor cells, respectively, in response to ionizing radiation [28, 29]. Though these and other inhibitory ligands are also markers of T-cell activation, T cells upregulate these ligands and receptors upon activation as a physiologic check on the immune system to avoid autoimmunity—possibly explaining the rarity of clinically relevant abscopal effects and may offer rational targets for combinatorial approaches using radiation therapy. Numerous preclinical studies demonstrate that RT is synergistic with checkpoint-blocking therapies [30], though clinical success has been limited [31]. So far, most published studies have targeted CTLA-4, but there are encouraging preclinical studies combining radiation with blockade of PD-1 and other key checkpoints and immunosuppressive pathways (reviewed extensively by Kang et al. [32]).
Finally, radiation is highly lymphotoxic, hence its continued use in some stem cell transplant-conditioning regimens for lymphoid leukemias. Thus, depending on dose and the scope of the treatment field, RT risks eliminating the very T cells the treatment aims to activate. This supports the use of limited field and hypo-fractionated dosing in order to achieve the optimal vaccinal effect; preclinical models have supported the benefit of hypo-fractionated dosing in optimizing antitumor immune responses [33]. A comprehensive review of radiation as an adjuvant to emerging immunotherapies is provided in this series by Professors Formenti and Demaria, leaders in this field.
Though radiation and chemotherapy are both typically considered lymphotoxic, chemotherapy can also serve multiple roles as an adjuvant to immune therapy. Like radiation, chemotherapy causes immunogenic tumor cell death resulting in the release of both immunogenic ligands and tumor antigen that can be taken up by intratumoral APCs as well as those in the surrounding tissue and draining lymphatics, extensively reviewed by Kepp et al. [34]. Aside from tumor cytotoxicity, lymphotoxic chemotherapies such as cyclophosphamide have been found to preferentially deplete regulatory T cells (Tregs), with a milder effect on T effector cells [35].
The role of chemotherapy in inciting the initial step of priming an immune response, as well as the role of old and new cytotoxic chemotherapies in combination with emerging immunotherapies, is reviewed elsewhere in this series by Professor Kroemer, an authority on immunogenic cell death.
28.3 Recruitment of Immune Infiltrate
Though immunogenic cell death is likely necessary to initiate and potentiate a tumor-specific immune response, the rarity with which the abscopal effect is observed, and the paucity of studies demonstrating that radiation or chemotherapy increases tumor-specific T cells, suggests that additional signals are needed to potentiate the anti-TAA immune response. To promote antigen uptake, one extensively used approach has been the addition of growth factors inducing APC differentiation and recruitment into tumor tissue. In contrast to systemic cytokine administration, direct intratumoral injection of cytokines and growth factors has also been tried and showed promise in augmenting the response to immune-based therapies, including granulocyte-macrophage colony-stimulation factor (GM-CSF) and FMS-like tyrosine kinase 3 ligand (Flt3L).
Preclinical studies have assessed the immunostimulatory capabilities of multiple cytokines and growth factors by transducing tumors with the corresponding genes in a murine melanoma model. These studies looked at a panel of genes including IL-2, IL-4, IL-5, IL-6, IFNγ, IL1-RA, ICAM, CD2, TNF-α, and GM-CSF; GM-CSF achieved the most profound tumor rejection and induction of tumor-specific CD4+ and CD8+ T cells which protected mice from rechallenge with non-transduced melanoma cells [37]. GM-CSF is a hematopoietic growth factor, and outside the marrow compartment, GM-CSF can be produced by lymphocytes and myeloid lineage cells, as well as non-hematopoietic cell types within the tumor microenvironment including fibroblasts, endothelial cells, and epithelial cells; GM-CSF is capable of activating both innate and adaptive immune response, and expression is typically promoted by inflammatory cytokines such as IL-6 and TNF-α, while immunosuppressive cytokines such as IL-10 decrease its expression [38–42]. Clinically, autologous tumor cells from 21 melanoma patients were transduced with GM-CSF, and subsequent tumor biopsies revealed greater T-cell infiltration, as well as tumor necrosis, fibrosis, and edema, in the majority of patients [43]. Comparable immune responses were seen in a subset of patients treated in similar trials of autologous GM-CSF-transduced tumor cells from patients with melanoma, as well as lung and prostate cancer [44–46]. Combinations of GM-CSF with treatments that promote immunogenic cell death have also demonstrated induction of antitumor immunity. One ISV trial in follicular lymphoma combined local radiotherapy and intratumoral rituximab with intratumoral injection of immature autologous dendritic cells and GM-CSF; this permutation resulted in an overall response rate (ORR) of 36%, and those in whom there was a measurable tumor-directed CD8+ response had the most profound and durable clinical responses [47].
Though attraction and maturation of DCs lead to a pro-inflammatory immunogenic signal, intratumoral GM-CSF has also been found to be a chemoattractant to neutrophils and other myeloid lineage cells capable of promoting angiogenesis and suppressing CD8+ T-cell priming. GM-CSF furthermore directly promotes the growth of some tumors by enhancing expression of matrix metalloproteinases (MMPs) instrumental to growth and migration and metastases [48]. Some human and murine cancers have been shown to constitutively express GM-CSF, or in some cases G-CSF, as well as the respective receptor, leading to autocrine promotion of proliferation [49–55]. Some propose that the role of GM-CSF may differ based on the tissue involved, such that in some cancers, its use may be an immune adjuvant, and in others it may promote tumor growth and suppress the immune response. A large cohort of gene arrays of human colorectal cancers demonstrate that over a third of cancers produce GM-CSF, and colorectal cancer patients with an increased level of GM-CSF in serum have been shown to have a worse prognosis [55, 56]. In head and neck cancer, GM-CSF and other growth factors in the serum were associated with worse invasion and prognosis overall [57, 58]; similarly, invasive bladder cancers occasionally secrete GM-CSF, and some patients’ cancer cells also express the GM-CSF receptor, and both these findings correlate with worse prognosis [53, 59]. Leukocytosis and elevated GM-CSF levels in blood or in tumor samples are seen in a variety of other cancers as well, including lung and glioblastoma multiform, some of which are the targets in immunotherapy trials incorporating GM-CSF [54, 60, 61]. These data prompt consideration of alternate growth factors capable of differentiating or recruiting APCs to the tumor site.
Flt3L is a hematopoietic growth factor crucial to the mobilization and differentiation of stem cells and progenitors [62]. Flt3L has been shown to facilitate the differentiation of both DCs and NK cells from hematopoietic stem cell precursors [63–66]. Specifically, Flt3L directly induces the differentiation of a proportion of CD34+ CD45RA-early progenitor cells into a DC precursor and results in increased numbers of plasmacytoid DC (pDC) and CD141+ and CD1c+ (cDCs) [63, 67]. These DC subsets, particularly the CD141+ subset, are especially capable of cross-presentation of tumor antigen to CD8+ T cells, and their intratumoral presence correlates strongly with clinical outcomes [68, 69].
Numerous preclinical studies have demonstrated the ability of Flt3L to mobilize immune cell subsets, DCs predominant among these [67, 69–73], including models showing antitumor effect of Flt3L administration in melanoma, lymphoma, leukemia, and breast, colon, prostate, lung, and hepatocellular carcinoma [74–82]. Other models have demonstrated that single-agent Flt3L is ineffective, rather the optimal antitumor immune response requires combinatorial approaches which induce antigen release and stimulation of APCs; for instance, in a murine model of breast adenocarcinoma, Flt3L priming potentiated the abscopal effect elicited by low-dose radiation and eliminated systemic tumor burden in a T-cell-dependent fashion, while Flt3L alone had no effect without radiotherapy [83].
Clinically, a phase I study evaluated the use of autologous DCs that first were expanded in vivo with Flt3L, loaded ex vivo with carcinoembryonic antigen peptide, and reinfused; treatment was well tolerated and demonstrated that Flt3L expanded DCs 20-fold; 2 of 12 patients experienced dramatic tumor regression [84]. Clinical response correlated with the expansion of CD8+ CEA tetramer+ T cells, confirming the role of CD8+ T cells in this treatment strategy. Similar robust increases in intratumoral and systemic DCs were seen in patients in additional early phase trials with peritoneal carcinomatosis, mesothelioma, and prostate cancer, all with limited toxicity [85, 86]. These early phase trials suggested that Flt3L is able to significantly increase the presence and activity of intratumoral APCs and that—similar to GM-CSF—it appears that combinatorial approaches are necessary to prime clinical remissions. Indeed, while Flt3L administration promotes DC production systemically and intratumoral infiltration, these DCs are immature DCs, which lack costimulatory molecules needed to activate effector cells. This both limits their ability to promote an effector T-cell response and actually promotes tolerogenic pathways keeping tumor-specific T cells quiescent [73]. As such, though Flt3L appears to augment the number of intratumoral DCs, additional pro-inflammatory stimuli are necessary to activate these APCs upon arrival.
28.4 Activation of Dendritic Cells
Single-agent studies of GM-CSF and Flt3L have demonstrated low toxicity but also low efficacy, confirming what we know about the physiological role of DCs in priming and homeostasis; it is not enough to promote the ingress of APCs into a tumor; an activation signal is needed. While this can come from DAMPs released by immunogenic cell death, arguably the strongest innate immune responses can also be elicited by introduction of pathogen-associated molecular patterns (PAMPs). TLRs are evolutionarily conserved innate immune receptors capable of recognizing PAMPs, as well as some DAMPs, and likewise TLR ligands, such as lipopolysaccharide (LPS), free single- or double-stranded RNA (ssRNA or dsRNA), and hypomethylated DNA motifs found in pathogens, are able not only to bind and activate canonical TLR pathways but also other cytoplasmic inflammatory pathways [87]. The sum stimulus of these pathways results in a maturation signal potentiating DC antigen presentation and cross-presentation and subsequent T-cell activation.
TLRs are resident either on the cell surface (as is the case for TLRs 1, 2, 4, 5, and 6) or in the endosome (TLRs 3, 7, 8, and 9), and their distribution is key to the PAMP and DAMP ligands they recognize. TLR7 classically recognizes free ssRNA, and agonists for this TLR were the first PAMP analog to be FDA approved for clinic use in the form of imiquimod. TLR7 is highly expressed on multiple myeloid lineages, with highest expression on plasmacytoid DCs that play a key antiviral role through induction of type I IFNs (IFNα/β). Imiquimod is an imidazoquinoline derivative capable of activating TLR7 and induces a strong antiviral immune response; it was first approved for the treatment of genital warts caused by a cutaneous viral infection and subsequently for the treatment of basal cell carcinoma and actinic keratosis, with off-label indications for cutaneous viral infections such as molluscum contagiosum [88–90].
Preclinical studies have also investigated the use of imiquimod in other cancers that are not virally derived. In a murine model of cutaneous breast cancer, topical application of imiquimod combined with local radiotherapy resulted in complete regression of locally treated tumors with abscopal responses at distal sites suggesting induction of T-cell-mediated antitumor immunity [91]. In this model, response is associated with increase in T-cell infiltration into tumor lesions, and the abscopal effect was dependent on CD8+ T cells. Similar trials have been performed in small series or case reports in patients with cutaneous involvement of metastatic melanoma or breast cancer; topical application of imiquimod induces a pro-inflammatory tumor microenvironment and some tumor regressions [92–94]. There are also many series or case reports of antitumor effects of imiquimod in cutaneous T-cell lymphomas [95–103] and B-cell lymphomas [104–107], now a common off-label use of this adjuvant.
In a combinatorial approach using imiquimod and a human cancer-testis antigen NY-ESO-1 peptide vaccine in melanoma, patients were pretreated with Flt3L, and imiquimod was applied to the vaccine sites [108]. Unfortunately, clinical response rates to this vaccine triplet regimen were suboptimal, and only 12 of 27 patients were evaluable, a minority of whom were treated with imiquimod, and only one patient experienced a partial response.
Systemic treatment with imiquimod has been assessed in preclinical models, with oral treatment leading to significant IFN induction and tumor growth inhibition in colorectal, sarcoma, and lung cancer models [109]. In a model of lymphoma, intravenous delivery of R848, a guanosine derivative which acts as an agonist for both TLR7 and TLR8 (another endosomal TLR which recognizes ssRNA), combined with local tumor irradiation induced durable CD8+ T-cell antitumor immune responses, clearance of systemic disease, and protection from tumor rechallenge [110]. A novel imidazoquinoline, 3 M-052 or MEDI9197, which is also a TLR7 and TLR8 ligand formulated with a lipid tail to allow tissue retention, was shown in preclinical models to suppress tumor growth following intratumoral injection both at target and distal lesions [111]. This antitumor effect was enhanced by PD-L1 and CTLA-4 blockade, even in models in which checkpoint blockade alone was ineffective. A phase I basket trial of intratumoral MEDI9197 injections in metastatic disease is underway, with recent cohorts combining MEDI9197 with anti-PDL1 therapy, hoping to reproduce the preclinical synergistic activation of antitumor immune responses [112].
There has been extensive study of the role of type I IFNs in the control of tumor growth, and these IFNs appear integral to mount an immune response to tumors [113, 114]. Preclinical studies suggest that this is due not to the role of IFNs in stimulating the cytotoxic T cells but also the role IFNs has in inducing maturation of DCs, particularly BATF3+ CD8α+ DCs in mice (the corollary of CD141+ DCs in humans which also express the BATF3 and IRF8 characteristic transcription factors) which are the most capable of cross-presenting tumor antigen to CD8+ T cells [115, 116].
TLRs are potent stimuli of IFNs; however, there are also cytoplasmic PRRs capable of inducing immune responses. One of these keys to the induction of the IFNs is the STING receptor, a receptor for cGAMP, a degradation product from cytoplasmic DNA [117]; STING activation has been shown to strongly activate type I IFN transcription [118]. Preclinical studies of intratumoral injection of a STING agonist DMXAA (also known as ASA404) into murine B16 melanoma were sufficient to achieve priming of tumor-specific CD8+ cells and achieve immune rejection of the tumor [119, 120]. DMXAA was tried in conjunction with chemotherapy in a large phase III trial of NSCLC in humans with no appreciable activity, though that was later determined to be due to poor binding to human STING [121]. ADU-S100 is a novel cyclic dinucleotide that binds and activates both human and mouse STING [122], and this is currently being used intratumorally in early phase trials.
It has also been noted that cancer cells may downregulate the STING pathway in order to avoid induction of IFN in response to DNA damage commonly present in rapidly dividing cancer cells [123]. This has been exploited by the field of oncolytic viruses using either pathogenic viruses that have been engineered to decrease virulence in normal tissue (such as herpes simplex virus, HSV, and vaccinia) [123, 124] or viruses that are typically not virulent in human cells but are able to infect and propagate in cancer cells in which the IFN pathway has been silenced (such as Newcastle disease virus, NDV) [125]. Oncolytic viruses are not only capable of inducing tumor lysis and antigen release; the viral components can activate innate PRRs. Initial limited clinical efficacy of these viruses alone made it evident that an additional immune stimulus such as a growth factor or cytokine could potentiate the immunity induced by viral infection. JX-594, a vaccinia virus, was engineered to express GM-CSF in infected cells, has been shown to selectively replicate in tumor cells, and demonstrated antitumor immunity in preclinical models and early phase clinical studies after either intratumoral or systemic inoculation with virus [124]. These studies revealed increased infiltration of T cells into the tumor and disruption of tumor-associated vasculature in mice and humans, leading to reduced blood flow and rapid necrosis of tumor cells [126–128]. This effect is tumor-specific due to loss of normal IFN response, as endothelial cells of normal blood vessels are not affected. Clinical trials in liver cancer and melanoma have found JX-594 is well tolerated and response rates have been encouraging [129–133]. Similarly, HSV, engineered to not express genes that block antigen presentation on MHC molecules and other genes key to viral replication in normal cells, demonstrated significantly enhanced immunogenicity by inserting GM-CSF into the viral genome [134]. Clinical trials of this construct referred to as talimogene laherparepvec or T-VEC (previously OncoVEX GM-CSF) have shown potent induction of necrosis in treated tumors and inflammation of distal tumors and significant response rates including complete responses to single-agent therapy in patients with melanoma, head and neck cancer, and breast and other metastatic malignancies [135–138]. A phase III study of T-VEC in patients with advanced melanoma demonstrated superior response rates compared to GM-CSF alone and a trend toward improvement in overall survival (p = 0.051), which led to approval of T-VEC for clinical use [139]. Combinations of oncolytic virus with immunomodulators such as checkpoint-blocking antibodies offer great promise; these trials (e.g., NCT03069378, NCT02965716) and the field of oncolytic viruses in general are reviewed elsewhere in this series by Professor Puzanov.
While STING is a cytoplasmic PRR recognizing free DNA, endosomal TLR9 is another PRR capable of sensing free DNA, specifically DNA containing hypomethylated CG-enriched oligonucleotide (CpG) islands commonly seen in the prokaryotic genome. TLR9 is expressed by pDCs and other DC subtypes to varying degrees and is also expressed on B cells, which can also be effective APCs. Interestingly, B-cell lymphomas are derived from mature B cells expressing high levels of TLR9, and CpG containing ligands can activate lymphoma B cells, just as they can with nonmalignant B cells, to increase expression of costimulatory molecules [140–142]. In a lymphoma model, CpG ligands inhibited proliferation of malignant B cells, and when combined with systemic chemotherapy capable of inducing immunogenic cell death, intratumoral CpG induced antitumor response and disease regression at site of injection as well as distal lesions in a CD8+ T-cell-dependent fashion, suggesting CpG was potentiating priming of tumor-reactive T cells [142]. While tumor-resident DCs may also respond to TLR9 ligands, in this model, this effect was seen in mice with somatic TLR9 deficiency, carrying TLR9-expressing tumors, suggesting that antigen presentation by tumor cells, not host APC, may be sufficient to induce potent immune responses. The TLR9 agonist agatolimod (PF-3512676) has been used in clinical trials in humans, including three trials using a combination of local irradiation and intratumoral agatolimod administration which achieved clinical responses, including some complete responses, among the 45 patients with indolent B-cell lymphoma and 15 patients with mycosis fungoides that were treated [143–145].
CpG has been shown to also elicit tumor regression in non-B-cell malignancies; in a murine glioma model, intratumoral CpG increased T-cell infiltration, eliminated tumors, and protected from rechallenge [146], and similar antitumor T-cell-mediated responses are primed with CpG injection in murine models of mesothelioma, breast cancer, and melanoma [147–149]. In humans, another TLR9 agonist, litenimod (CpG-28), was given as an intratumoral injection into 34 patients with recurrent glioblastoma multiforme, achieving some partial responses [150]. The above CpG molecules are classified as CpG type B (CpG-B) in that they optimally activate B cells, whereas a CpG-C molecule SD-101—designed to comparably activate B cells and pDC—has shown promising preclinical and clinical results upon intratumoral administration and is currently being studied in lymphomas and solid tumors including combination studies with anti-PD1 antibodies (NCT02521870, NCT02731742, NCT02927964, NCT03007732).
As noted above, the importance of CD141+ DCs in the tumor relates to their potent ability to cross-present antigen and prime a potent CD8+ T-cell response to tumor antigen. It is noteworthy that these cells express very low levels of TLR7, TLR8, and TLR9; however, they express high levels of TLR3, another PRR located in the endosome which responds to dsRNA [151]. Polyinosinic-polycytidylic acid stabilized with polylysine and carboxymethyl cellulose (poly-ICLC, also known as hiltonol) is a stabilized double-stranded RNA (dsRNA) “host-targeted” therapeutic viral mimic that exhibits broad innate and adaptive immune-enhancing effects and was shown to be a promising vaccine adjuvant due to its strong induction of type I IFNs [152, 153]. As such, it was first used as an IFN inducer at high doses in cancer trials, with predictably high toxicity [154], while at lower doses, it achieved a broader host defense stimulation, potent adjuvant effect, and antiproliferative and antiviral effects [155–157].
The possible antitumor and antiviral activity of poly-ICLC is thought to be dependent on its stimulation of both TLRs and MDA5 (another cytoplasmic PRR [158]) and its concomitant induction of IFNs and other cytokines and its induction of MHC molecules. Likewise, dsRNA induction of various costimulatory factors such as B7-H2, CD40, and OX40 may help overcome evasions in the tumor itself [159].
Administration of poly-ICLC has been demonstrated to prolong survival in multiple murine tumor models. In a murine lymphoma model, IP administration of poly-ICLC significantly prolonged survival and correlated with peritoneal macrophage tumoricidal activity [160]. Poly-ICLC also prolonged survival in rodent models of gliomas, melanoma, and fibrosarcoma [161, 162]. Poly-ICLC incorporated into peptide-based vaccine for gliomas showed ability to induce tumor lymphocyte infiltration and induction of an IFN signature [163–165], and poly-ICLC used as an adjuvant with the E7 HPV tumor antigen in a murine model of cervical cancer demonstrated expansion of antigen-specific CD8+ T cells as well as tumor regression [166]. While single-agent poly-ICLC can induce tumor responses, our group has developed a combined approach ISV in the murine lymphoma A20 model in which poly-ICLC is used to activate intratumoral DCs after treatment with low-dose radiation and intratumoral Flt3L (Fig. 28.2 is the human correlate). While this approach does induce tumor regression and cure in approximately one-third of animals, we have shown that addition of checkpoint blockade with an anti-PD-1 mAb results in regression of nearly all tumors, further supporting the importance of both inducing intratumoral inflammation and modulating the immunosuppressive tumor microenvironment to achieve the optimal antitumor immune response [167].
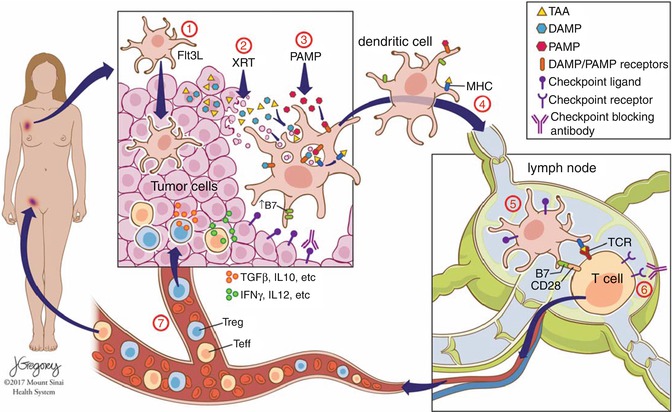
Fig. 28.2
Novel ISV protocol. Intratumoral Flt3L (1) induces recruitment of DCs to the tumor, which subsequently take up antigen after low-dose radiation (2), and DCs are activated by intratumoral injection of PAMPs such as poly-ICLC (3), inducing activation of antigen-loaded DCs, which then migrate to the draining lymph nodes (4) where they interface with tumor antigen-specific T cells (5). Activation threshold of senescent or exhausted tumor-specific T cells can be overcome through addition of checkpoint blockade (6), and activated T effector cells then travel to tumor sites throughout the body (7) to exert systemic antitumor effect and promote tumor-specific memory
Poly-ICLC has been used in dozens of clinical trials in multiple solid malignancies and lymphoma which have been reported, and many more ongoing clinical trials in combination with chemotherapies, radiation, and/or other immune adjuvants, with varying clinical success, but with promising data, demonstrated the ability to elicit immune response to tumor antigen [165, 168–178]. Based on these and other studies in humans, poly-ICLC is thus emerging as a particularly potent immunogenic core for multiple cancer and HIV vaccines, given its adjuvant role in potentiating antigen uptake, cross-presentation, and skewing of immune response to tumor antigens toward Th1 phenotype [176]. A trial of 22 patients with recurrent malignant gliomas used autologous DCs loaded ex vivo with synthetic TAA peptides, administered with concurrent intramuscular poly-ICLC, resulting in potent antigen-specific T-cell responses, and a suggestion of prolonged survival [179]. A similar peptide-based vaccine trial in patients with advanced ovarian cancer used a vaccine that combined overlapping long peptides from NY-ESO-1 in conjunction with poly-ICLC; while the peptide alone induced no immune response, the investigators saw significantly higher induction of NY-ESO-1-specific antibodies as well as antigen-specific T-cell responses in 10 of 11 patients that received the poly-ICLC [174]. Another vaccine in multiple tumor types expressing NY-ESO-1 attached full-length NY-ESO-1 protein to DCs by conjugating the tumor antigen to an antibody for DEC-205 expressed on DCs; this antibody-peptide conjugate was administered in conjunction with poly-ICLC, again this adjuvant was found to be well tolerated, and a small minority had robust clinical regression of metastatic disease [180]. Another study of the cancer testes antigen MAGE-A3 similarly treated patients with whole peptide and poly-ICLC, followed by autologous stem cell transplant and adoptive transfer of ex vivo expanded T cells, and found that over three quarters of the 25 patients treated developed cellular and humoral antigen-specific responses [173].
Intratumoral injection of poly-ICLC to achieve an in situ vaccination is currently being studied as well, alone and in combination. In an ongoing trial of single-agent intratumoral poly-ICLC used in patients with metastatic malignancies, a patient with advanced facial rhabdomyosarcoma had significant intratumoral inflammation and necrosis followed by marked tumor regression [175]. In an ongoing study aimed at potentiating the abscopal response, intratumoral injection of recombinant Flt3L and poly-ICLC is administered in combination with low-dose radiotherapy; early reports demonstrate increased intratumoral DCs and regression of both treated tumors and systemic disease in some patients with advanced-stage follicular lymphoma [181, 182]. Given recent preclinical results demonstrating the potency of this approach in combination with checkpoint blockade, the upcoming iteration of this (intratumoral) ISV will study the combination along with (systemic) anti-PD1 mAb (Fig. 28.2).
28.5 Modulating Immunosuppressive Cellular Compartment
While most of the monotherapy and combinatorial approaches we have covered thus far are able to induce tumor-specific T cells and responses in a subset of patients, many patients remain refractory to these therapies. This is likely due to the T-cell response working in opposition to other driving immune force within the tumor microenvironment, e.g., subsets of myeloid and lymphoid cells exerting a strong immunosuppressive effect. Through secretion of immunosuppressive chemokines and cytokines as well as direct cell contact, malignant cells promote the development and recruitment of immunosuppressive Tregs, tumor-associated macrophages (TAMs), and MDSCs [183–186]. Along with immunosuppressive costimulatory ligands and cytokines expressed by tumor cells, these leukocytes act in concert to limit immune reactivity to tumor cells; to effectively induce an immune response against tumor antigen, this immunosuppressive milieu will likely need to be disrupted (Fig. 28.3).
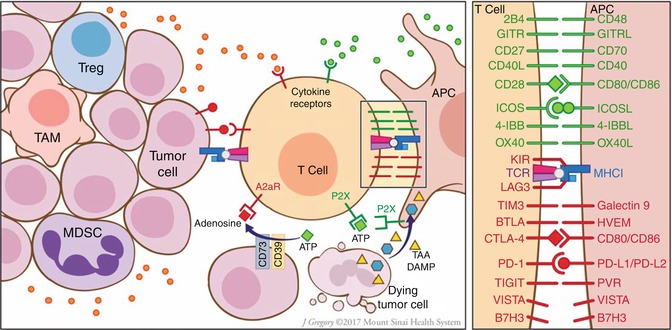
Fig. 28.3
Modulating the stimulatory/inhibitory components of the tumor microenvironment. The interface between the T cell and APC (right panel), which in many ways mirrors the ligand-receptor interactions seen between the T cell and the tumor cell, offers a multitude of targetable costimulatory and inhibitory ligands. Furthermore, tumor cells and recruited immunosuppressive cells such as Tregs, TAMs, and MDSCs promote immune evasion through release of cytokines skewing the effector response and through conversion of activating DAMPs such as ATP into inhibitory adenosine
A critical cell responsible for modulating effector T-cell responsiveness is the Treg, and there is extensive literature demonstrating that malignant cells secrete chemokines recruiting intratumoral Tregs, which act to impair proliferation of T effector cells and impair their degranulation and cytotoxic activity [187, 188]. As mentioned previously, in addition to their cytotoxic effect on cancer cells, certain chemotherapies such as cyclophosphamide, anthracyclines, and platinum agents have been shown to exert preferential cytotoxicity to Tregs over effector T cells [15, 189]. In patients treated with chemoimmunotherapy, one study demonstrated a rapid reconstitution of the CD8+ and NK cell number and function, while the CD4+ compartment—particularly Tregs—remained depleted for up to a year [190]. While chemotherapy could play a role in conditioning before vaccination, most of the agents may also impede optimum lymphocyte responses, so to deplete Tregs in a more targeted manner, some vaccine trials have incorporated daclizumab, a humanized anti-CD25 antibody which depletes Tregs [191–193]. While the ultimate clinical effect in these early phase trials was negligible, the treatment effectively depleted Tregs in the circulation and could be considered for use in conjunction with other combinatorial immunotherapy-vaccine approaches.
Similar to tumor cells, TAMs and MDSCs express high levels of suppressive surface antigens including PD-L1 and through cytokines and direct modulation promote senescence of intratumoral T cells or skew the phenotype away from optimal cytotoxic effector functions; their presence and prevalence are unsurprisingly associated with a poor prognosis [194, 195]. While there is a paucity of antigen-specific surface markers on MDSCs and TAMs that would allow for targeting, Bruton’s tyrosine kinase (BTK) is expressed in MDSCs, and treatment of mouse and human MDSCs with the Tec-kinase inhibitor ibrutinib significantly impaired cell migration and production of immunosuppressive cytokines [196, 197]. Though developed to target BTK, ibrutinib acts on other Tec-family kinases such as the IL-2-inducible T-cell kinase (ITK), which can also skew T-cell maturation toward an effector phenotype, further countering the immunosuppressive microenvironment [198]. This blockade of immunosuppressive pathways was further shown in a recent preclinical study of CpG-based ISV with adjuvant ibrutinib that demonstrated enhanced antitumor response [199]. Ibrutinib is now being used in combination with a variety of immunomodulatory compounds including checkpoint-blocking antibodies, with promising clinical responses.
There are also promising clinical and preclinical studies of other immunomodulatory compounds such as lenalidomide that suggest a possible adjuvant role in vaccine protocols. One preclinical trial looking at direct immunosuppressive activity of cancer cells found that tumor-infiltrating lymphocytes (TILs) developed impaired immune synapse formation with lymphoma cells upon incubation with malignant cells, blocking cytotoxic activity of the T cells, and that this defect in synapse formation was reversed after treatment with lenalidomide [200]. Recent studies of lenalidomide combined with rituximab in the treatment of lymphoma found increased ADCC-mediated killing by NK cells [201], and trials in multiple myeloma demonstrated improved efficacy of PD-1 blockade when given concomitantly with lenalidomide [202]. Clinical trials using lenalidomide with a variety of immunotherapies within and outside the hematological compartment are ongoing.
Conclusion
The field of cancer immunotherapy is rapidly evolving, with many new checkpoint-blocking and agonistic antibodies in the clinical trial pipeline. While these offer great promise, we know from studies of CTLA-4 and PD-1 blockade that inflamed tumors are most responsive to immune-mediated therapies. As such, local immunotherapies, such as ISV approaches that focus on homing APCs to a tumor, and stimulating immunogenic cell death and inflammatory pathways alongside checkpoint agonist/antagonist mAbs, tyrosine kinase inhibitors, or other immunomodulatory drugs will likely yield the rational combination therapies that allow us to achieve better clinical response rates and more durable immune memory.
References
1.
Busch W. Aus der Sitzung der medicinischen section vom 13 November 1867. Berl Klin Wochenschr. 1868;5:b99.
2.
Coley WB. The treatment of malignant tumours by repeated inoculations of erysipelas with a report of ten original cases. Am J Med Sci. 1893;105:487.
3.
Otto F, et al. Phase II trial of intravenous endotoxin in patients with colorectal and non-small cell lung cancer. Eur J Cancer. 1996;32A:1712–8.PubMed
4.
Goto S, et al. Intradermal administration of lipopolysaccharide in treatment of human cancer. Cancer Immunol Immunother. 1996;42:255–61.PubMed
5.
Engelhardt R, Mackensen A, Galanos C. Phase I trial of intravenously administered endotoxin (Salmonella abortus equi) in cancer patients. Cancer Res. 1991;51:2524–30.PubMed
6.
Ansell SM, et al. Phase I study of ipilimumab, an anti-CTLA-4 monoclonal antibody, in patients with relapsed and refractory B-cell non-Hodgkin lymphoma. Clin Cancer Res. 2009;15:6446–53. doi:10.1158/1078-0432.CCR-09-1339.PubMedPubMedCentral
7.
Ansell SM, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372:311–9. doi:10.1056/NEJMoa1411087.PubMed
8.
Armand P, et al. Programmed death-1 blockade with pembrolizumab in patients with classical hodgkin lymphoma after brentuximab vedotin failure. J Clin Oncol. 2016; doi:10.1200/JCO.2016.67.3467.
9.
Zinzani PL, et al. Phase 1b study of PD-1 blockade with pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma (PMBCL). Blood. 2015;126:3986.
10.
Spranger S, et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell–inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci. 2016;113:E7759–68. doi:10.1073/pnas.1609376113.PubMedPubMedCentral
11.
Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv324. doi:10.1126/scitranslmed.aad7118.
12.
Hellmann MD, Gettinger SN, Goldman JW, Brahmer JR, Borghaei H, Chow LQ, Ready N, Gerber DE, Juergens RA, Shepherd FA, Laurie SA, Zhou Y, Geese WJ, Agrawal S, Li X, Antonia SJ. CheckMate 012: safety and efficacy of first-line (1L) nivolumab (nivo; N) and ipilimumab (ipi; I) in advanced (adv) NSCLC. J Clin Oncol. 2016;34(15):3001.
13.
Sharma, P. in Society for the Immunotherapy of Cancer.
14.
Wolchok JD, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33. doi:10.1056/NEJMoa1302369.PubMedPubMedCentral
15.
Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. doi:10.1146/annurev-immunol-032712-100008.PubMed
16.
Mole RH. Whole body irradiation; radiobiology or medicine? Br J Radiol. 1953;26:234–41. doi:10.1259/0007-1285-26-305-234.PubMed
17.
Apetoh L, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–9. doi:10.1038/nm1622.PubMed
18.
Ghiringhelli F, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–8. doi:10.1038/nm.2028.PubMed
19.
Obeid M, et al. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ. 2007;14:1848–50. doi:10.1038/sj.cdd.4402201.PubMed
20.
Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5. doi:10.1038/nature00858.PubMed
21.
Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell. 2010;140:798–804. doi:10.1016/j.cell.2010.02.015.PubMed
22.
Deng L, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41:843–52. doi:10.1016/j.immuni.2014.10.019.PubMedPubMedCentral
23.
Matsumura S, et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol. 2008;181:3099–107.PubMedPubMedCentral
Related posts:
 Peptide-Based Therapeutic Cancer Vaccines
Peptide-Based Therapeutic Cancer Vaccines
 The Human Tumor Microenvironment
The Human Tumor Microenvironment
 Synergy Between Radiotherapy and Immunotherapy
Synergy Between Radiotherapy and Immunotherapy
 Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
 Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
 Immune Monitoring of Blood and Tumor Microenvironment
Immune Monitoring of Blood and Tumor Microenvironment
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


