The term lobular neoplasia (LN) encompasses the entire spectrum of atypical epithelial lesions that originate in the terminal duct-lobular unit (TDLU) of the breast, and are characterized by a population of dyshesive cells, which expand the lobules and acini of the TDLUs, and may involve the terminal ducts in a pattern known as Pagetoid spread.1 These lesions were traditionally described under the terms lobular carcinoma in situ (LCIS) and atypical lobular hyperplasia (ALH), which refer to the degree of involvement of the acinar structures of a given TDLU.
The first description of LCIS, as an “atypical proliferation of acinar cells” of the breast, was reported by Ewing in 1919.2 The main characteristics of this lesion, however, were not thoroughly documented until 1941 in the seminal study by Foote and Stewart.3 The term LCIS was chosen to emphasize the histologic similarities between the cells of LCIS and those of frankly invasive lobular carcinoma (ILC), and, importantly, was not meant to infer that the cell of origin resided in the lobules. In fact, it was acknowledged that LCIS would originate in the TDLU and small ducts. Based on the frequent identification of LCIS in association with ILC, and following the analogy of ductal carcinoma in situ (DCIS) and invasive ductal carcinoma (IDC), Foote and Stewart3 hypothesized that the neoplastic cells of LCIS would still be contained within a basement membrane, and that this lesion would constitute a precursor of breast cancer development, leading to the recommendation for mastectomy.
Emerging data throughout the 1970s from Haagensen et al4 and others5 demonstrated that the risk of breast cancer development following a diagnosis of LCIS was lower than that expected for a direct precursor lesion (approximately 1% per year) and was conferred equally to both breasts, generating controversy regarding the significance of LCIS and leading to disparate recommendations for management, ranging from observation only to bilateral mastectomy.
The term ALH was coined in 1978 to refer to a less prominent in situ proliferation composed of cells cytologically identical to those of LCIS which were associated with a significantly lower risk of subsequent breast cancer development; approximately one-half of the risk associated with LCIS.6 However, as the distinction between LCIS and ALH, which is based on quantitative rather than qualitative differences between the lesions (described below), often proves challenging in diagnostic specimens, Haagensen et al4 put forward the term “lobular neoplasia” to refer to the entire spectrum of these in situ lesions, including ALH and LCIS.
In addition to the classic forms of LN, several variants of LN have also been described. Of potential clinical significance is the pleomorphic variant (see below), first described in its pure form by Sneige et al7 under the name of pleomorphic lobular carcinoma in situ (PLCIS). Recognition of this variant is important, given that its histological features can lead to difficulty in differentiating between PLCIS and DCIS.
In current practice, a diagnosis of ALH or LCIS is typically perceived as a risk indicator rather than a precursor of subsequent carcinoma and, as such, radical treatment has fallen out of favor. Yet, observational evidence to suggest that the risk of breast cancer development following a diagnosis of LN is higher in the ipsilateral than in the contralateral breast,8 and compelling molecular data which demonstrate that ALH and LCIS are clonal neoplastic proliferations that commonly harbor the same genetic aberrations as those found in adjacent invasive cancers8–12 have reinstated the notion that ALH and LCIS are both nonobligate precursors and risk indicators of invasive breast cancer.
Lobular carcinoma in situ is most frequently diagnosed in women 40 to 55 years of age as an incidental microscopic finding in a breast biopsy or excision specimen obtained for another reason.3,4,13 The true prevalence of LCIS in the general population is difficult to estimate, and likely exceeds the incidence, given that it does not present as a mass lesion nor does it have a specific radiographic appearance. The reported incidence of LCIS in otherwise benign breast biopsy specimens ranges from 0.5% to 3.8%,4,13 whereas population-based data reported to Surveillance, Epidemiology, and End Results (SEER) from 1978 to 1998 demonstrate an incidence of 3.19 per 100,000 women,14 with the highest incidence rate (11.47 per 100,000 person-years) in 1998 among women 50 to 59 years of age.15
Histologically, LCIS is often multifocal and bilateral, with >50% of patients diagnosed with LCIS showing multiple foci in the ipsilateral breast, and bilateral lesions are reported in approximately one-third of patients.16,17 This pattern of presentation, combined with evidence of familial clustering,18,19 has led to the hypothesis that these lesions could be underpinned by germline genetic abnormalities. Data to support this, however, are limited to a small number of families with a hereditary form of diffuse gastric cancer and breast lobular carcinoma caused by CDH1 germline mutations.20 Outside of this context, the potential genes involved and the pattern of inheritance of familial LCIS remain uncertain.1
In its classic form, LN is characterized by variable enlargement and distention of the acinar structures by a neoplastic population of monomorphic, dyshesive, small, round, or polygonal cells, often with inconspicuous cytoplasm (Fig. 72-1). The lobular architecture is largely maintained, and the neoplastic cells display a regularly spaced distribution. Intracytoplasmic lumina and vacuoles, sometimes containing a central eosinophilic dot (known as magenta body), are common,1,3,4,21 as is pagetoid spread, whereby the neoplastic cells extend between the intact overlying epithelium and the underlying myoepithelial layer and basement membrane.
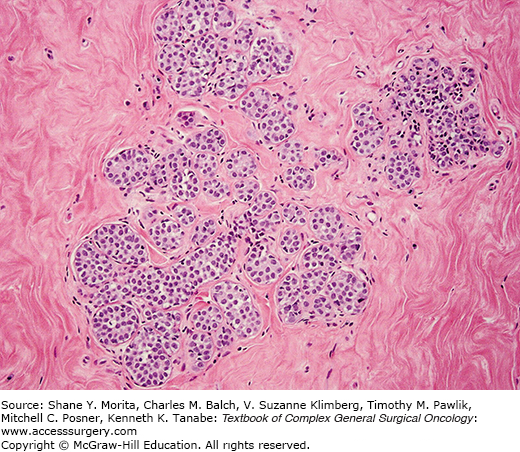
Some variability in the cytomorphology between cases, and frequently within the same case, may be appreciated, and two cytologic subtypes have been recognized. Type A cells are small, dyshesive cells, with scant cytoplasm and small, bland (often nuclear pleomorphism score of 1) nuclei (about 1.5× the size of that of a lymphocyte), whereas type B cells have more abundant, often clear cytoplasm, medium-sized nuclei (about 2× bigger than a lymphocyte), mild-to-moderate nuclear atypia (nuclear pleomorphism 1 or 2), and indistinct or absent nucleoli.7,21 This cytological classification scheme has neither clinical utility nor correlates with the risk of invasive breast cancer. It does, however, serve as a reminder that some degree of cytologic variation can be observed in bona fide cases of classic LN, and that moderate atypia does not warrant a diagnosis of PLCIS.
The subclassification of LN into ALH and LCIS is quantitative rather than qualitative.1 For a diagnosis of LCIS, more than half the acini in an involved lobular unit must be filled and distended by the characteristic cells, leaving no central lumina, whereas ALH is defined as a less well-developed and less-extensive lesion, where the characteristic cells only partly fill the acini, with minimal or no distention of the lobule.1,8,21,22 Although the use of this subclassification is justified on the basis of the lower risk conferred by ALH than by LCIS, the differentiation between ALH and LCIS based on these criteria is subjective and depends on the extent of sampling of a given lesion. For diagnostic purposes, the term LN rather than ALH or LCIS is recommended, particularly in core needle biopsy specimens.21,23
However, a more relevant distinction is between the classic form of LN and PLCIS.24 This variant is characterized by pleomorphic cells that are substantially bigger than those of classic LN,7,24 and by more abundant, pink, and often finely granular cytoplasm. Features of apocrine differentiation are frequently found.24,25 PLCIS nuclei are atypical, pleomorphic, and large (4× the size of lymphocyte nucleus), often containing conspicuous nucleoli (Fig. 72-2). PLCIS not uncommonly presents with central, comedo-type necrosis and microcalcifications; yet, necrosis is not required for the diagnosis. Recognition of the pleomorphic subtype is important because the combination of cellular features, necrosis, and calcification can lead to difficulty in differentiation from DCIS, and potentially overtreatment, although data regarding the natural history of PLCIS are very limited. Importantly, while some advocate for a more aggressive approach in the management of patients with PLCIS, with treatment recommendations akin to those for DCIS, this approach is only supported by molecular studies which have demonstrated that PLCIS shares many similarities with pleomorphic ILC, not by long-term outcome studies demonstrating the actual risk of subsequent cancer development.
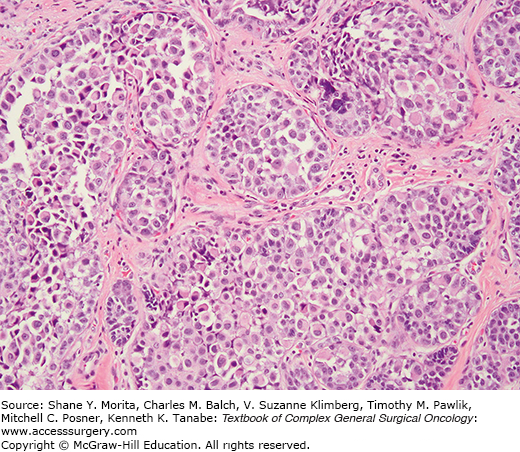
A further system for classification of LN has been proposed using the terminology lobular intraepithelial neoplasia (LIN), with subdivision, based on morphologic criteria and clinical outcome, into three grades (LIN 1, LIN 2, LIN 3), with LIN 3 representing the PLCIS end of the spectrum.26,27 This system presupposes that the risk of invasive carcinoma development would be related to increasing grade of LIN. This classification system, however, is supported by limited evidence and has not been endorsed in the latest WHO classification.1
Lobular neoplasia, in its classic forms, is typically characterized by strong expression of ER-alpha (ER⍺), ER-beta (ERß), and PR; low proliferation indices as defined by Ki-67, and lack of expression of HER2 and p53, features that are consistent with those of estrogen receptor (ER) positive breast cancers with a less-aggressive clinical behavior (i.e., luminal A subtype).11,28,29
The phenotypic characteristics of PLCIS are more varied; although the majority of these lesions do express ER and progesterone receptor (PR), their expression is usually at lower levels, and truly ER-negative cases of PLCIS have been documented, particularly those with apocrine features. HER2 gene amplification and HER2 overexpression can also be found in a subset of PLCIS, and intermediate-to-high Ki-67 labeling indices, usually higher than those of classic LCIS, are a common feature of these lesions.25,30 The apocrine subtype of PLCIS, which is composed of cells with overt apocrine cytology and which express GCDFP-15 (gross cystic disease fluid protein-15), a marker of apocrine differentiation,1,21,25 are frequently found to have HER2 gene amplification and high proliferation rates. However, it should be noted that the criteria to differentiate between PLCIS and apocrine PLCIS remain a matter of controversy.
One of the most frequent genetic aberrations in ER-positive breast lesions, particularly those of low histological grade, is 16q loss, which occurs in a high proportion of cases as an early event in the neoplastic development of LN and low-grade DCIS.1,9,11,12,21 While the target gene of 16q deletions in ductal lesions remains to be identified, in lobular lesions, the CDH1 gene, which encodes E-cadherin, has been shown to be the target.1,11,12,21
E-cadherin is a transmembrane adhesion molecule found in adherens junctions and mediates homophilic-homotypic adhesion in epithelial cells; its intracytoplasmic domain is bound to p120 catenin and ß-catenin. In breast epithelial cells, loss of E-cadherin results in cytoplasmic, and rarely nuclear, accumulation of p120 catenin, and in loss of ß-catenin membranous expression, but without nuclear accumulation of ß-catenin or activation of the canonical Wnt pathway.31,32 The mechanisms resulting in CDH1 gene silencing in LCIS, PLCIS, and ILC include a combination of genetic, epigenetic, and transcriptional mechanisms. Loss of 16q is usually accompanied by CDH1 inactivating mutations, CDH1 homozygous deletions, and CDH1 gene promoter methylation.1,11,21
Lack or marked downregulation of E-cadherin expression is observed in >95% of ALH, LCIS, PLCIS, ILCs, and metastatic deposits of ILCs, and has now been shown to be the cause of the characteristic dyshesiveness of LN and PLCIS cells . The study of other components of the E-cadherin-catenin complex in LN, PLCIS, and ILCs has revealed that these lesions are also characterized by lack of ß-catenin membranous expression and cytoplasmic expression of p120 catenin.
The study of CDH1 gene mutations in ALH, LCIS, and synchronous ILC has provided direct evidence to suggest that some LN and ILCs are clonally related, given the presence of identical CDH1 gene mutations in the LN and ILC components.1,11,12,21
Genome-wide genetic analyses of gene copy number aberrations and allelic changes found in LN have revealed that these lesions are clonal and neoplastic; their most frequent copy number changes include 16p, 16q, 17p, and 22q, and gain of material from 6q.1,9,11,25,33,34 These analyses have also demonstrated that classic LCIS and a substantial proportion of adjacent synchronous lesions, including ER-positive DCIS, ILC, and ER-positive invasive ductal carcinoma, are often clonally related.9,34 Similar observations have been made through the analysis of mitochondrial DNA heteroplasmy and mitochondrial gene mutations,10 supporting the contention that some LN are nonobligate precursors of more advanced ER-positive lesions (e.g., DCIS, invasive lobular, and invasive ductal carcinomas).
Related posts:
 Defining the Specialty of Surgical Oncology
Defining the Specialty of Surgical Oncology
 Medullary Thyroid Cancer
Landmark Clinical Trials that Impacted Surgical Management of Invasive and Noninvasive Breast Cancer
Palliation Of Incurable Gastric Cancer
Medullary Thyroid Cancer
Landmark Clinical Trials that Impacted Surgical Management of Invasive and Noninvasive Breast Cancer
Palliation Of Incurable Gastric Cancer
 Cystic Lesions of the Liver and Biliary Tract
Cystic Lesions of the Liver and Biliary Tract
 Skin Closure After Resection Of Skin Malignancies, Including Melanoma
Skin Closure After Resection Of Skin Malignancies, Including Melanoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


