Age at diagnosis (years)
15–19
20–24
25–29
US population (in millions), year 2000 census
19.90
18.70
17.63
Average incidence, 1975–2000, per million
2.0
5.6
14.6
Average annual increase, 1975–2000, SEER
0
1.8
2.6
Estimated incidence, year 2000, per million
2.0
5.6
14.6
No. of persons diagnosed with liver and intrahepatic bile duct cancer, year 2000, USA
41
105
258
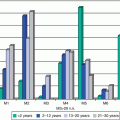
The incidence of liver cancer is relatively constant between 5 and 35 years of age, but then it steadily increases with age (Fig. 18.1). Available SEER data collected between 1975 and 2011 have shown a steady increase in the incidence of liver and intrahepatic bile duct tumors between 1976 and 2011 for all age groups and for both sexes (Figure Incidence Trend). The distribution of different histologic types varies with age. While hepatoblastoma accounts for 90 % of liver tumors in children less than 5 years of age and less than 5 % for those older than 15 years of age, hepatocellular carcinoma presents two peaks of incidence: the first during the early AYA years (age 15–29) accounting for 80 % of the liver tumors in this age group and the other in the middle-aged population. Hepatic sarcomas are a rare type of liver tumors that occur primarily before 15 years of age, while adenocarcinoma of the liver, cholangiocarcinoma, and other intrahepatic bile duct tumors peak during the AYA age range (Figs. 18.2 and 18.3 Biopathology by Age). Eleven percent of the liver tumors in the AYA population originate from the intrahepatic bile duct.
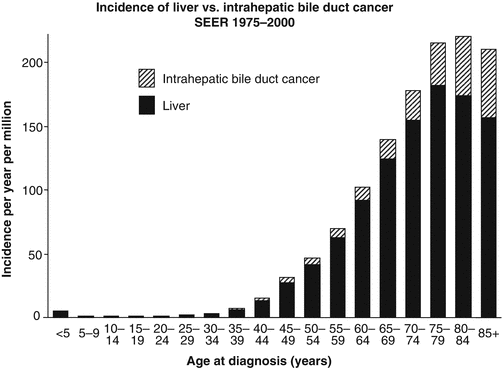
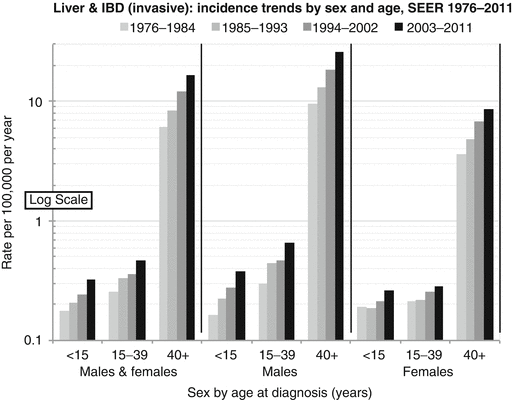
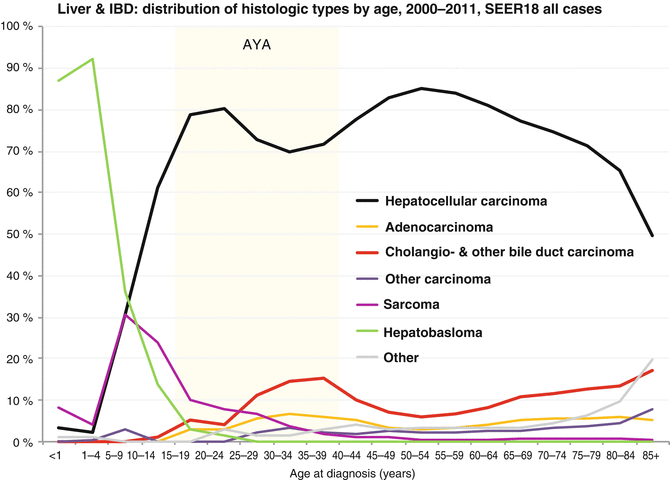
Fig. 18.1
Relative incidence during 1975–2000 of liver and intrahepatic bile duct cancer in US patients by age at diagnosis (Data from the US SEER Program (Bleyer et al. [1]))
Fig. 18.2
Incidence of liver and intrahepatic bile duct cancer in the USA by sex and age. IBD intrahepatic bile duct (Data from the US SEER Program Bleyer WA; Source: SEER 9 Areas, NCI)
Fig. 18.3
Histologic types of liver and intrahepatic bile duct cancer by age. IBD intrahepatic bile duct (Data from the US SEER Program Bleyer WA; Source: SEER 18 Areas, NCI)
The incidence of liver cancer is greater in males than females in all age groups (Fig. 18.4 Incidence). Liver tumors are more prevalent in Asians/Pacific Islanders in AYA and older persons (Figs. 18.4, 18.5, 18.6, and 18.7 Incidence).
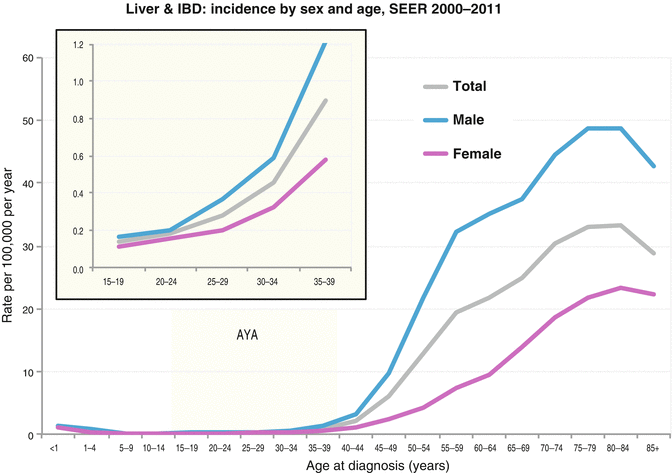
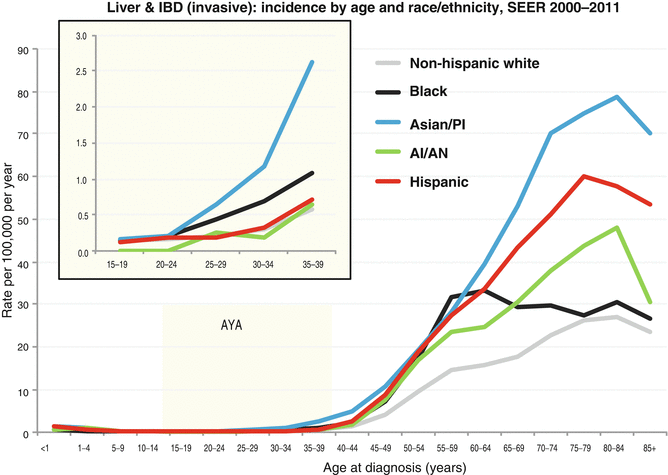
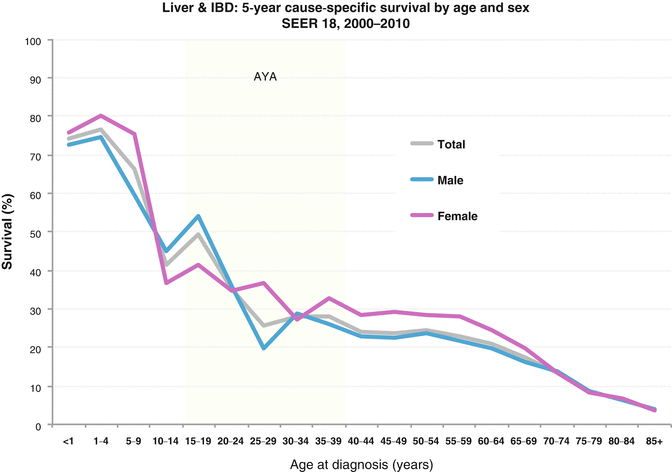
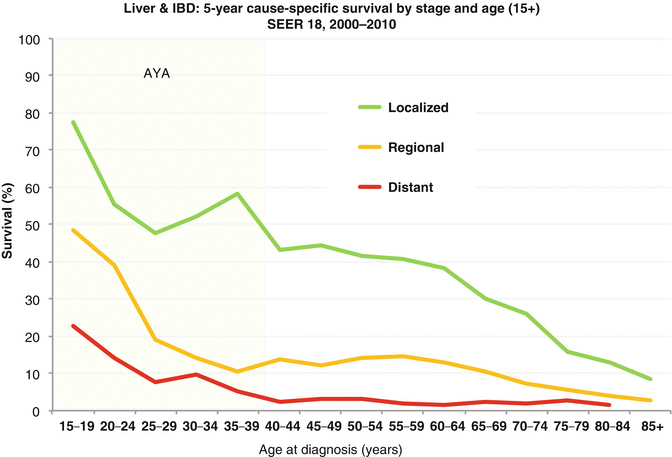
Fig. 18.4
Incidence of liver and intrahepatic bile duct cancer in the USA by sex and age, 2000–2011. IBD intrahepatic bile duct (Data from the US SEER Program Bleyer WA; Source: SEER 18 Areas, NCI)
Fig. 18.5
Incidence of liver and intrahepatic bile duct cancer in the USA by age and race/ethnicity, 2000–2011. IBD intrahepatic bile duct (Data from the US SEER Program Bleyer WA; Source: SEER 18 Areas, NCI). Note: Hispanic may overlap with black, Asian/PI (Asian/Pacific Islander), and AI/AN (American Indian/Alaska Native)
Fig. 18.6
5-year survival of patients in the USA with liver and intrahepatic bile duct cancer by age and sex, 2000–2011. Abbreviations: IBD intrahepatic bile duct (Data from the US SEER Program Bleyer WA; Source: National Cancer Institute: SEER18 Areas)
Fig. 18.7
5-year survival of patients in the USA with liver and intrahepatic bile duct cancer by stage and age, 2000–2011. Abbreviations: IBD intrahepatic bile duct (Data from the US SEER Program Bleyer WA; Source: National Cancer Institute: SEER18 Areas). Excludes unstaged – too few cases
Bosch et al., utilizing population-based cancer registries and the World Health Organization (WHO) mortality data bank, reported the incidence and mortality of liver cancers worldwide [2]. With an estimated 437,000 new cases in 1990, liver cancers ranks fifth in frequency in the world, accounting for 5.4 % of all human cancer cases. Liver cancer corresponds to 7.4 % of all cancer cases among men and 3.2 % of all cancers among women. The largest estimated concentration of liver cancer cases are located in East Asia (China, Hong Kong, Korea, Mongolia, and Japan), in Middle Africa (Cameroon, Chad, Congo, and Equatorial Guinea), and in some Western African countries (Gambia, Guinea, Mali, and Senegal [3]). The lowest concentration of liver cancer is seen in Northern Europe, in Australia, in New Zealand, and in the Caucasian populations in North and Latin America [3].
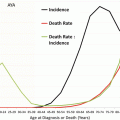
Liver cancer-specific survival rate decreases steadily with age. Survival in AYA is one-half that seen in young persons, and for persons 25 years or older, it is similar to those of middle-aged adults (Fig. 18.6 Survival). For all ages, disease presentation with regional or distant extension is associated with worse outcomes. While the 5-year liver cancer-specific survival rate for AYAs with localized disease at diagnosis was approximately 50 %, those with regional or distant involvement continue to have a dismal outcome with survival rates less than 20 % (Fig. 18.7 Survival). The overall death rate from liver and intrahepatic bile duct cancer has steadily increased since the early 1980s. The death rate for AYAs has been relatively constant to slightly improve since the early 1980s; however, that due to intrahepatic bile duct cancers has dramatically increased over the years (Fig. 18.8 Death Rates).
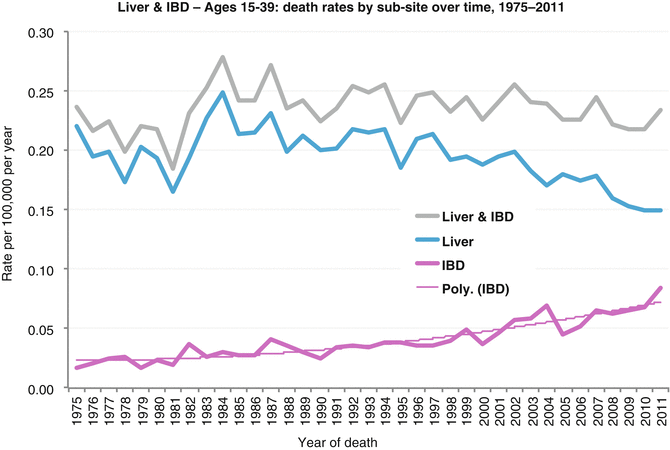
Fig. 18.8
Death rate of patients in the USA with liver and intrahepatic bile duct cancer (ages 15–39) by tumor site, 1975–2011 (Data from the US SEER Program Bleyer WA; Source: NCHS Mortality, Total U.S.)
18.3 Risk Factors and Etiology
Hepatocellular carcinomas appear to result from complication of previous hepatic damage due to metabolic or inflammatory disorders. Chronic infection with hepatitis B virus is the leading cause of HCC in children, adolescents, and young adults in Asia and Africa. However, in the Western countries, less than a third of the adolescent or young adult patients diagnosed with HCC have an identifying cause such as hepatitis or other inflammatory liver diseases [4, 5]. This is in marked contrast to older adults in which almost 90 % of the cases are cirrhosis related, secondary to viral infection or alcohol consumption [6]. The prevention of a carrier state in children by a universal program of hepatitis B immunization has shown a dramatic decrease in the chronic hepatitis B virus prevalence and a decline in the rates of HCC in Taiwan among children less than 15 years of age [7].
Less frequently HCC is associated with congenital diseases such as hereditary tyrosinemia, biliary cirrhosis, glycogen storage disease, and alpha-1 antitrypsin deficiency [8–11]. Prolonged exposure to anabolic steroids, toxin-contaminated foods (aflatoxin), and potential hepatic carcinogens (pesticides, vinyl chloride, thorotrast) have also been associated with the development of HCC [12–14].
18.4 Pathology and Biology
18.4.1 Hepatocellular Carcinoma (Adult Type)
The pathologic features of hepatocellular carcinoma (HCC) of the adult type (ordinary HCC) are well established (Figs. 18.9). Adult-type HCC also develops in children, adolescents, and young adults. Similar to cancers occurring in the older age group, HCCs in younger individuals display distinct macroscopic growth patterns, including expanding, invasive, pedunculated, multinodular, and diffuse lesions. The main clinical and biological features have been reviewed [15–17]. To date, no differences have been recognized among children, adolescents, and adults in the biology and pathology of typical, ordinary HCC. A systematic analysis of the significance of histopathologic risk factors identified in patients with adult-type HCC [18] has yet to be performed in adolescents and young adults.
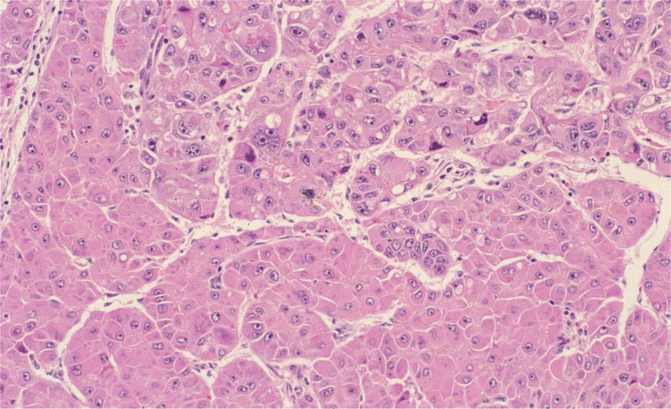
Fig. 18.9
Hepatocellular carcinoma, trabecular type: thick plates without typical intervening sinusoids
18.4.2 Fibrolamellar Hepatocellular Carcinoma
Fibrolamellar hepatocellular carcinoma (FL-HCC) is a primary liver cancer characterized by a single expanding mass with typical histologic features consisting of large eosinophilic cells embedded in an abundant collagen-rich stroma, the latter forming “fibrolamellae” between the cancer cells (Fig. 18.10). This tumor occurs most often in young individuals. It accounts for about 30 % of HCC in patients younger than 20 years of age. Typically, FL-HCC develops in the absence of underlying cirrhosis, hepatitis viral infection, or metabolic disorders, and serum AFP is not elevated. FL-HCC may be associated with high serum vitamin B12-binding capacity, express a neuroendocrine signature [20], and exhibit aromatase activity causing gynecomastia [21]. A clear cell variant of FL-HCC exists [22]. The neoplasm has distinct immunohistochemical features [23], including reactivity for cytokeratin 7 [24]. FL-HCC markedly differs from ordinary HCC with respect to karyotypic and genomic abnormalities. Recently, a recurrent and highly characteristic chimeric transcript located to chromosome 19 has been identified in FL-HCC. The transcript encodes a protein containing the amino-terminal domain of DNAJB1, a homolog of the molecular chaperone DNAJ, fused in frame with PRKACA, the catalytic domain of protein kinase A (DNAJB1-PRKACA fusion) [25]. The fusion results in increased cAMP-dependent protein kinase A activity, and the fusion protein is oncogenic in HCC cells. Later studies confirmed the association of this chimeric transcript with FL-HCC, suggesting that this alteration is a highly characteristic molecular signature for FL-HCC [26–29]. A second gene fusion event found in FL-HCC is a translocation between CLPTM1 and GLIS3 genes, generating a transcript that promotes malignant transformation in cell lines [29].
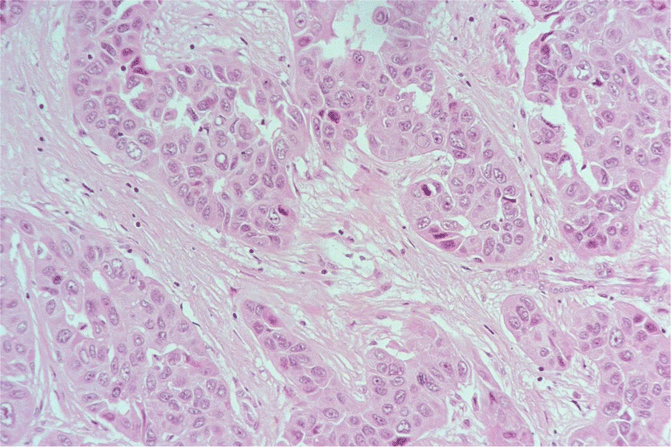
Fig. 18.10
Fibrolamellar carcinoma: solid nests of large and eosinophilic cells are embedded in a collagen-rich stroma forming fibrolamellar structures
18.4.3 Hepatoblastoma
Hepatoblastoma (HB) is rare in adolescence and adulthood [30]. In fact, the large majority of HB is diagnosed in infants and children less than 5 years old. There is evidence that HB is the most prominent member of a HB family of tumors, which share distinct molecular features, including abnormalities of the Wnt/beta-catenin signaling pathway (see below). Conventionally, HBs were divided into epithelial HB and mixed epithelial-mesenchymal HB, the latter subclassified into those with or without teratoid features [31–33] (Figs. 18.11). A novel classification was worked out in the frame of an International Pathology Symposium in March 2011 in Los Angeles. Twenty-two expert pathologists of COG, SIOPEL, GPOH, and JPLT as well as pediatric oncologists and surgeons formulated a consensus classification published in 2014 [34] (Table 18.2).
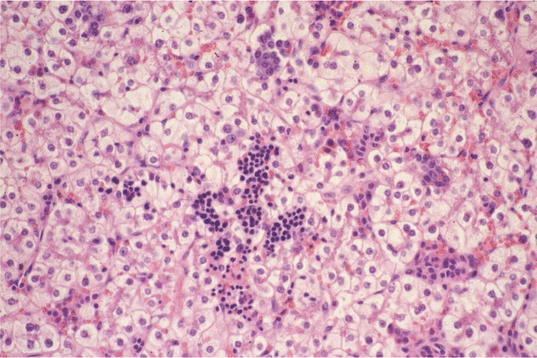
Fig. 18.11
Hepatoblastoma fetal morphology type: most of the tumor cells exhibit a clear cytoplasm. Extramedullary hemopoiesis is present
Table 18.2
Los Angeles hepatoblastoma classification
Epithelial hepatoblastomas |
Fetal HB: |
Well differentiated: uniform (10–20 μm), round nuclei, cords with minimal mitotic activity (<2 mitotic figures per 10/400× microscopic fields); extramedullary hematopoiesis present |
“Crowded” or mitotically active: more than two mitotic figures per 10/400× microscopic fields; conspicuous nucleoli, usually less glycogen |
“Pleomorphic or poorly differentiated”: moderate anisonucleosis, high nucleus/cytoplasmic ratio, and prominent nucleoli |
“Anaplastic”: marked nuclear enlargement and pleomorphism, nuclear hyperchromasia, and abnormal mitotic figures |
Embryonal HB: |
Tumor cells 10–15 μm in diameter, high nucleus/cytoplasmic ratio, angulated nuclei, primitive tubular structures, and extramedullary hepatopoiesis usually absent |
Macrotrabecular: |
Epithelial HB (fetal or embryonal) growing in trabeculae of more than five cells thick (between the sinusoids) |
Small cell undifferentiated: |
Tumor cells 5–10 μm in diameter, no distinct architectural pattern, minimal pale amphophilic cytoplasm, round to oval nuclei with fine chromatin structure and inconspicuous nucleoli, and mitotic figures present; part of tumors lack nuclear INI1 expression |
Cholangioblastic HB: |
Bile duct-like profiles are present, usually at the periphery of epithelial HB islands; this pattern may predominate |
Mixed epithelial–mesenchymal hepatoblastomas |
HB with stromal derivatives: |
Presence of spindle cells (“blastema”), osteoid, skeletal muscle, and cartilage |
Teratoid HB: |
Mixed HB plus primitive endodermal components, neural derivatives, melanocytes, and glandular elements |
HB can develop in young adults and older patients [35]. The pathology of HB is the same in young and older subjects, and the criteria for histological diagnosis have been reviewed recently [32, 36]. However, molecular signatures will be required to test whether adult HBs are the same or different entities in comparison with HB of young age. There are intriguing situations whereby HCC occurs in combination with HB [37] or HCC recurring as HB [38], suggesting a possible filiation of these neoplasms. Part of hepatic tumors in older children and adolescents contain HCC-like components and exhibit signs of cholangiocyte differentiation. Such highly aggressive lesions have been termed “transitional liver cell tumor” [39] and may be related to pleomorphic or anaplastic variants of HB. Similar to infants and children, adolescents and young adults may also develop unusual HB family tumors that have recently been identified, including small cell HB with “rhabdoid features” (tumors that do not immunohistochemically express the chromatin-remodeling protein INI1/hSNF5/SMARCB1) [40], and cholangioblastic HB.
The oncogenic pathways of HCC and HB differ in many respects. HCCs show multiple chromosomal aberrations (mainly losses), whereas HBs exhibit a lower number of chromosomal changes [30, 41]. In contrast to HCC, p53 gene (and related genes) mutations are almost lacking in HB, and p53 protein overexpression is seen infrequently [15, 42–44]. HB can develop in the setting of imprinting disorders, including Beckwith-Wiedemann syndrome (BWS) involving the loss of imprinting of IGF2 [45]. In BWS, HB was only detected in patients with chromosome 11p15 paternal uniparental disomy (UPD) [46]. HB can also complicate paternal uniparental disomy 14 and epimutations and microdeletions that affect the maternally derived 14q32.2 imprinted region, a constellation found in Kagami-Ogata syndrome [47].
In cancerogenic pathways leading to HB, abnormalities of the Wnt/beta-catenin signaling pathway play a central role [41, 48]. This signaling cascade is critically involved in cancer stemness and malignant behavior [49]. Beta-catenin gene mutations in HB involve the degradation targeting box [50], and the activation of beta-catenin involves both epithelial and mesenchymal HBs [51]. Mutations of beta-catenin gene in HB are associated with overexpression of proliferation factors, including cyclin D1 [52]. Based on its role on priming and expansion of stem and progenitor cells, beta-catenin activation may affect hepatic stem cells, including DLK1-positive oval cells present in HB [53]. Beta-catenin activation in a distinct progenitor cell type is sufficient to cause HB and HCC [54]. Beta-catenin can activate different transcriptional programs in subsets of HB, associated with distinct expression of hepatic stem-cell markers in immature tumors [55]. Alterations of other components of the Wnt signaling pathway have been found in HB, including mutations of the adenomatous polyposis coli (APC) and AXIN genes.
A further hepatic tumor showing abnormal Wnt/beta-catenin signaling is nested stromal-epithelial tumor (NSET; ossifying stromal-epithelial tumor) [56, 57]. This neoplasm occurs in both the pediatric age group and in adolescents and rarely in adults and is characterized by multiple clustered nests of immature epithelial cells embedded in a spindle-cell stroma with osteoid foci, surrounded by a sheath of myofibroblasts. NSET may be associated with Cushing’s syndrome due to ectopic ACTH production [58] and usually shows a benign course, although recurrent and metastatic disease occurs. Marked nuclear reactivity of nested cells for beta-catenin is a consistent finding [31]. A later study uncovered mutations of the beta-catenin gene in NSET, characterized by large deletions in exon 3, suggesting that this neoplasm is related to HB with defective mesenchymal-epithelial transition [59]. This relation is supported by the association of NSET with Beckwith-Wiedemann syndrome [60]. Beta-catenin mutations occur in a subset of hepatocellular adenoma. Beta-catenin-activated hepatocellular adenoma can give rise to malignant hepatic tumors, including well-differentiated atypical HCC [61].
Recently, gene mutations affecting cellular functions other than those linked to Wnt signaling were detected in HB, including the ubiquitin ligase complex [62] and the transcriptional coactivator, Yap1 [63]. A subset of HB exhibits mutations of the transcription factor NFE2L2, involving residues that are recognized by the KEAP/CUL3 complex for proteasomal degradation [64]. Aggressive subsets of HB with HCC-like properties showed loss of genomic stability and promoter mutations of telomerase reverse transcriptase (TERT) [64].
18.5 Clinical Presentation and Diagnosis
Since HCC in adults most often presents on a background of chronic liver disease, the symptoms are frequently masked by those associated with the underlying disease and are frequently found during routine screening exams in at-risk individuals. In children and adolescents with no underlying liver disease, the symptoms are usually of short duration, and most often patients present with an enlargement of the abdomen and an associated palpable right upper quadrant mass. Anorexia, weight loss, and abdominal pain are frequently seen in association with advanced disease. Rarely, it may present as an acute abdominal crisis secondary to tumor rupture. Jaundice, vomiting, fever, and pallor are rare. On physical examination hepatomegaly is common, and a palpable hard mass is frequently found. If the tumor is associated with preexisting inflammatory or metabolic diseases of the liver, signs associated with cirrhosis of the liver can be found, including splenomegaly and spider angiomata. Most frequently there is extensive involvement of the liver by the tumor, and often the tumor is multifocal in origin. The presence of ascites may suggest intra-abdominal extension, and at least one-thirds of the patients present with metastatic involvement, with the lungs being the most common site of disease.
Mild normochromic-normocytic anemia can be seen, as well as thrombocytosis and occasionally polycythemia secondary to extrarenal secretion of erythropoietin. Hepatic enzymes can be elevated; however, elevation of bilirubin is infrequent, unless it is associated with cirrhosis of the liver.
Alpha-fetoprotein (AFP) is the most valuable laboratory test for diagnosis and monitoring of hepatic tumors. Alpha-fetoprotein is a normal globulin present during fetal life, synthesized in the liver and fetal yolk sac. Elevated levels of AFP are seen during the newborn period, and adult levels are reached by about 1 year of age. The biologic half-life of AFP is 5–7 days. The level of AFP at diagnosis has been shown to be of prognostic value, and it can be utilized to monitor response to therapy and disease recurrence in hepatoblastomas [65]. Alpha-fetoprotein levels, however, can be normal in at least 30–50 % of the patients with HCC. Levels of carcinoembryonic antigen (CEA) and ferritin can also be increased in hepatocellular carcinoma [66]. The fibrolamellar variant of hepatocellular carcinoma can be associated with an abnormality of the vitamin B12-binding protein, which can occasionally be used to monitor disease status and response to therapy [67]. Screening for viral hepatitis (B and C) should be performed in all patients.
Plain radiographs of the abdomen frequently demonstrate the presence of a right upper quadrant mass, and calcifications may be noted in approximately 6 % of the malignant tumors [68]. Ultrasonography is a reliable and noninvasive imaging technique in establishing the presence of an intrahepatic mass and when used in conjunction with AFP measurements is a sensitive tool for screening of patients at high risk of developing HCC. It aids in differentiating solid from cystic masses and in determining the presence and extension of vascular extension [69, 70]. HCC are highly vascular tumors that preferentially supplied by hepatic artery branches rather than the portal venous system [71]; therefore, when triple-phase imaging techniques are used, these tumors typically demonstrate contrast enhancement in the arterial phase and washout of contrast media in the portal venous phase. In the USA, computed tomography (CT) or magnetic resonance imaging (MRI) is currently the preferred modalities for imaging these tumors [72]. Despite the tremendous advancement in radiographic imaging, including [18F] fluorodeoxyglucose-positron emission tomography (FDG-PET) and diffusion MRI [73], changes in unidimensional tumor size, as evaluated by the Response Evaluation Criteria in Solid Tumors (RECIST) system, continue to be the most widely used method to assess treatment response [74]. There are numerous limitations in using the RECIST system for the evaluation of response, including the lack of reproducibility and the fact that size alone does not capture the biologic effects of targeted treatment.
18.5.1 Differential Diagnosis
Some other liver tumors (non-HB and non-HCC) which can occur in this age group should be considered in the differential diagnosis are discussed below.
18.5.1.1 Embryonal (Undifferentiated) Sarcoma of the Liver
It is a specific well-described but rare tumor not to be confused with rhabdomyosarcoma but generally responding to some similar type of chemotherapy used in the treatment of rhabdomyosarcoma [75].
It occurs mostly in older children and adolescents. Twenty-five percent occur between the age of 11 and 20 and 6 % between 16 and 20 years.
The tumor mainly presents as a large solitary mass often proceeded by rather unspecific abdominal symptoms. Liver function is usually not compromised.
The imaging can be confusing in that on ultrasound it appears solid but on CT and MRI may show cystic elements even so far as to be misinterpreted as a solitary cyst [76]. Therefore histological diagnosis is essential and can show some specific cells, i.e., “polygonal” cells.
There is no standard treatment protocol, and initially this tumor was considered highly malignant with a poor prognosis. This opinion has of late needed revising especially since the advent of preoperative chemotherapy [77]. Most tumors respond very well to rhabdomyosarcoma-like therapy, i.e., VAC regimen with vincristine, actinomycin, and cyclophosphamide, and to agents like doxorubicin and cisplatin. When these tumors are completely resected, the prognosis is relatively good [77].
With this approach and some personal experience, even some ruptured tumors are curable [78].
18.5.1.2 Adenocarcinoma, Cholangiocarcinoma, and Other Bile Duct Carcinomas
Since the incidence of adenocarcinoma, cholangiocarcinoma and other intrahepatic bile duct cancers peak in the AYA years they should be considered as part of the differential diagnosis.
18.5.1.3 Benign Tumors
Focal Nodular Hyperplasia (FNH) and Liver Cell Adenoma
These are essentially tumors of adults most commonly found in women taking contraceptives but occasionally also occur in the age group under consideration here, but with no know hormonal etiology. In the AFIP (Armed Forces Institute of Pathology Monograph (1977–1999)), 20 % of the total of the benign liver tumors seen between the age of 11 and 15 years were FNH.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


