Fig. 2.1
Neutrophil emigration into the alveolar air space. The thick alveolar wall is the preferred site for neutrophil emigration. (1) Initial mechanical sequestration followed by prolonged retention of a neutrophil within the lumen of a pulmonary capillary. (2) Trans-luminal crawling of a neutrophil towards the junction of endothelial cells. (3) Paracellular trans-endothelial migration of a neutrophil. (4) Neutrophils migrate through the holes in the endothelial basement membrane (thick black line) followed by migration through the ECM along the interstitial fibroblast and finally into the basal-lateral space beneath the type-II pneumocyte through the holes in the epithelial basement membrane (thick black line). (5) Neutrophils enter the alveolar lumen preferentially at the junction of type I and type II pneumocytes
The structure of the pulmonary microcirculation has been described elegantly in the recent review by Townsley et al. [31]. A single alveolus is surrounded by ~1000 inter-connected short tubular capillary segments arranged in a spheroidal mesh. The alveolar wall separating adjacent alveoli has a single network of capillaries which is exposed to the alveolar air on both sides [1, 32]. The average length and diameter of pulmonary capillary segments in the human lung is 15 and 7.5 μm, respectively, and the entire pulmonary capillary network is connected to 300 million arterial and venular end branches [2, 10, 29]. The lumen of the pulmonary capillaries is lined with pulmonary endothelial cells and a single endothelial cell is estimated to cover the walls of three adjacent capillary segments [33]. Figure 2.2 shows an MPE-IVM fluorescence microscopy image of the lung microcirculation in a wild type (WT) C57BL/6 mouse following intravascular infusion of FITC-dextran (green). As shown in Fig. 2.2, the terminal pulmonary arteriole branches out into networks of inter-connected short capillary segments, which surround alveoli (visible as dark polygons) and drain into the pulmonary venule (not visible in Fig. 2.2). These terminal arterioles and venules have a diameter in the range of 20–30 μm [13, 16, 19]. Also the physiological pressure difference across the whole pulmonary capillary network (between a pulmonary arteriole and venule) is ~6 mmHg, while that across a single pulmonary capillary segment is 0.15–0.6 mmHg [34, 35]. Thus, the pressure across a single pulmonary capillary segment is 10 times smaller than that across the whole pulmonary capillary network.
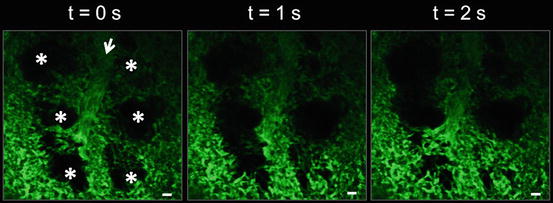
Fig. 2.2
Structure of the pulmonary microcirculation in mice. MPE-IVM fluorescence image of the pulmonary microcirculation in a WT C57BL/6 mouse following IV infusion of FITC-dextran (green). The pulmonary capillaries surrounding the alveoli (*) can be seen moving in and out of the imaging plane in the z-direction due to the dynamic expansion and contraction of the alveoli (*) with mechanical ventilation. Movement of the alveoli starts from the bottom of the image at t = 0 s and moves up to the top with increasing time. Alveoli are marked by asterisks. Intravascular FITC dextran highlights the pulmonary capillaries and a feeding arteriole in green. The open arrow denotes the direction of blood flow within the feeding arteriole. The times displayed are relative to the frame at t = 0 s. Scale bars are 20 μm. The feeding arteriole has a diameter of 33 μm, while the capillaries have an average diameter of 6 ± 2 μm. Figure adapted from Ref. [16]
Leukocyte Kinetics in the Noninflamed Lung
As described in the section “The blood–air barrier in the lung,” the average diameter of a pulmonary capillary segment in the human lung is ~7.5 μm, which is smaller than the average diameter of a human neutrophil ~ 8 μm [2, 4, 7, 36]. Therefore, neutrophils are required to deform in order to transit through the pulmonary capillary network (capillary network between a pulmonary arteriole and a venule). Based on in vivo studies performed in canine or rabbit lungs [7, 8, 21, 37], a neutrophil is estimated to pass through 40–100 capillary segments during a single transit from an arteriole to a venule and 30–60 % of these segments require the neutrophil to deform. Studies using scanning electron microscopy of fixed human lung tissue [7] and in vivo video microscopy of canine lungs [11] have revealed that neutrophils which are spherical in the pulmonary arterioles appear to be elliptical in shape inside the pulmonary capillaries. Recently, these findings were confirmed by high-resolution MPE-IVM studies performed in mice [13, 16], which revealed ellipsoid neutrophils slowly transiting through the capillary segments in the mouse lung (Fig. 2.3).
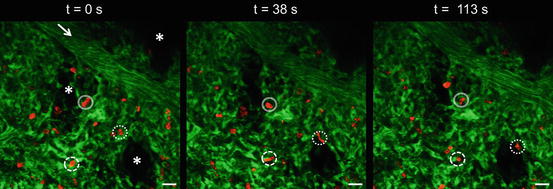
Fig. 2.3
Neutrophil kinetics in the noninflamed lung. MPE-IVM fluorescence image of the pulmonary microcirculation in a WT C57BL/6 mouse. Intravascular FITC dextran (green) highlights the pulmonary capillaries and a feeding arteriole. The arrow denotes the direction of blood flow within the feeding arteriole. Neutrophils shown in red were stained by IV infusion of Alexa Fluor 546 Gr-1 mAb (red). The marginated pool of neutrophils is visible as elliptical cells (red) within the pulmonary capillary segments (green). The solid and dotted circles denote three elliptical neutrophils slowly transiting through the pulmonary capillaries over a 2 min observation period. The times displayed are relative to the frame at t = 0 s. The pulmonary arteriole has a diameter of 30 μm. Alveoli are marked by asterisks. The scale bars are 20 μm
Deformation of neutrophils from a spherical to an elliptical shape is not instantaneous [38, 39]. Neutrophils are known to pause for a while at the entrance, as well as at the capillary junctions to deform, and the pause time is referred to as the “entrance time” [4, 7]. Computational models of the pulmonary capillary network, which were inspired by in vivo video microscopic images of rabbit and canine lungs, reveal that the pressure drop across a single pulmonary capillary segment needs to exceed a critical value to allow the cell to enter a capillary segment [34, 35]. This critical pressure is determined by the ratio of the capillary to neutrophil diameter [34, 38] and the cortical tension of the neutrophil ~35 pN/μm [39], which is the surface tension that enables the cell to maintain a spherical shape [40]. The cortical tension is dictated by the presence of a layer of actin cytoskeleton known as the “cortex” beneath the plasma membrane [39, 41]. Using in vitro pulmonary capillary sized microfluidic channels, Gabriele et al. [42] have shown that the entrance and transit time of passive leukocytes through 4 μm capillaries is determined by the degree of actin polymerization and therefore, stiffer cells take more time to enter into a capillary segment. In vivo, the pressure drop across a capillary segment can be smaller than the critical pressure; however, the blockage of the capillary entrance by a neutrophil can result in a 100–300 % increase in the pressure drop, which eventually pushes the neutrophil through the capillary [35].
Once inside a capillary segment, neutrophils also stop in the straight segments between junctions and these straight segment pauses are four to five times more frequent than in the junctions [11, 16]. The hemodynamic force acting on a stationary neutrophil that completely fills the lumen of the capillary segment can be estimated F≈ΔPπR2p, where ΔP is the pressure difference (~0.15–0.6 mmHg ) and R p is the radius of the capillary segment [36, 43, 44]. Based on IVM studies done in canine lungs, Lien et al. [9] showed that the RBCs being highly deformable, easily transit through a single pulmonary capillary network with a transit time of 1.4–4.2 s, while neutrophils being less deformable have a mean transit time of 6.1 min. The pauses responsible for the increased transit time of neutrophils result in at least a 50-fold higher concentration of neutrophils in the pulmonary than the systemic circulation and this resident pool of neutrophils is referred to as the “marginated pool” [4, 7, 21, 45]. The prolonged transit time and margination of neutrophils in the lung microcirculation also serves to promote host defense by allowing these cells to respond promptly to an inflammatory stimulus [10]. At the same time, the interconnected architecture of the pulmonary capillary network ensures that there are enough alternative routes for RBC transit around those capillary segments which are plugged with neutrophils [4].
Recent data from our lab (Fig. 2.3) and others [13, 28] using MPE-IVM of the lung in WT C57BL/6 mice revealed that the neutrophils are present in large numbers in the noninflamed lung, completely fill the lumen of the capillaries and therefore, appear ellipsoidal as they transit through the pulmonary capillaries [16]. Similar to previous reports [4, 11, 13, 28, 46], our analysis of neutrophil track lengths and speed revealed that the neutrophil transit through the pulmonary capillaries is indeed a combination of “hopping (stop and go)” and slow continuous motion. As shown in Figure 2.3, we found that some capillaries even in the noninflamed lung are effectively occluded temporarily as neutrophils transit slowly through them, which can take longer than 5 min as shown previously [35]. Although the ratio of neutrophil to capillary diameter and the forces of deformation are believed to dictate the transit time and margination of neutrophils within the noninflamed pulmonary capillary network, a role for neutrophil–endothelial adhesion cannot be entirely ruled out.
Neutrophil–endothelial adhesion plays a pivotal role in neutrophil margination within the systemic microcirculation. The neutrophil adhesion to the systemic venular endothelium starts with rolling, which is mediated by P-selectin on the endothelium binding to P-selectin-glycoprotein-ligand-1 (PSGL-1) on neutrophils [6]. Interleukin-8 (IL-8/CXCL-8) on human endothelium or KC (CXCL-1) on mouse endothelium binds to CXCR1 and CXCR2 on rolling neutrophils to activate β2-integrins CD11b/CD18 (Mac-1) and CD11a/CD18 (LFA-1) expressed on neutrophils, which then bind to intercellular-adhesion-molecule-1 (ICAM-1) on the endothelium to enable arrest [6]. Although endothelial P-selectin is central to leukocyte rolling in venules and arterioles [47], its role in leukocyte margination within the pulmonary capillaries is debatable [4]. To test the role of P-selectin (if present on the capillary wall) in mediating leukocyte margination in the lungs, undifferentiated HL-60 promyelocytic leukemia cells (diameter 13.5 μm) were allowed to flow in 7.5 μm diameter glass capillaries coated with recombinant human P-selectin or bovine serum albumin (BSA) under physiological conditions [43]. HL-60 cells were observed to deform into an elliptical shape and flow freely with minimal arrest in BSA coated capillaries but paused for long durations in P-selectin coated capillaries [43]. Although this may imply that P-selectin if present on the capillary endothelium can mediate neutrophil sequestration in pulmonary capillaries, there is conflicting evidence to support that such a scenario may exist in pulmonary capillaries in vivo [4]. P-selectin is expressed on the pulmonary arteriolar and venular endothelium [11, 48–50] and supports leukocyte rolling in these vessels in vivo [11, 47, 51]. However, the immunohistochemistry performed on noninflamed canine lung sections revealed that P-selectin is absent from the pulmonary capillary endothelium [11]. Alternatively, it has been proposed that the P-selectin expression on the pulmonary capillary endothelium can be low enough to be detected by the immunohistochemical techniques [2].
L-selectin is constitutively expressed on passive neutrophils and is known to mediate neutrophil margination in the systemic venules [52]; however, the requirement of L-selectin in neutrophil margination within the noninflamed lung is unclear. Using IVM of the rabbit lung under baseline conditions, Kuebler et al. [51] found that the intravenous infusion of fucoidin, which inhibits both P-selectin and L-selectin, led to the reduced rolling of leukocytes in both pulmonary venules and arterioles and also a reduction in the neutrophil transit time through the pulmonary capillary bed, suggesting a role for P- or L-selectin in leukocyte margination in the lung. A role for L-selectin was further supported by Gebb et al. [11], which used IVM to show that L-selectin mediates neutrophil rolling in the pulmonary arterioles and venules of noninflamed canine lungs. In contrast, Doyle et al. [53] revealed that the margination of neutrophils in pulmonary capillaries as well as venules and arterioles was normal in L-selectin deficient mice, suggesting that L-selectin may not be necessary for neutrophil margination in the noninflamed pulmonary capillaries.
Although β2-integrin-ICAM-1-mediated firm arrest is critical to neutrophil margination in the systemic microcirculation [6], both β2-integrin and ICAM-1 seem to be not required for neutrophil margination in the noninflamed lung. Aoki et al. [46] used IVM of the ex vivo perfused rat lung to show that ICAM-1 is constitutively expressed on both the pulmonary venular and capillary endothelium, suggesting that ICAM-1 may mediate neutrophil margination in the pulmonary capillaries. CD18 (β2-) integrins on circulating neutrophils are maintained in the bent passive state in the absence of inflammation and the binding of β2-integrins to ICAM-1 requires their activation to an extended high-affinity state, suggesting no role for β2-integrins in the noninflamed lung [54]. Anderson et al. [55] demonstrated that the neutrophil deformation followed by migration through a 3 μm polycarbonate filter resulted in the upregulation of CD18 expression on neutrophils, which led to their enhanced adhesion to the immobilized recombinant ICAM-1. Similar to this observation, other in vitro studies have shown that neutrophils activate following aspiration into glass capillaries smaller than 5 μm in diameter [43, 44]. Whether such deformation induced activation and upregulation of CD18 expression occurs in pulmonary capillaries in vivo is debatable [56] but the role of CD18 in neutrophil margination can be tested by inhibiting CD18 function. Animals injected with functional blocking antibodies (Abs) against CD18 showed no increase in circulating neutrophil count, suggesting that CD18 is not required for the margination of neutrophils in the noninflamed lung [4]. Finally, mice deficient in both P-selectin and ICAM-1 do not show a defect in neutrophil margination in the noninflamed lung and surprisingly, the size of the marginated pool in these double deficient mice was significantly larger than the WT mice suggesting that both endothelial P-selectin and ICAM-1 are not necessary for neutrophil margination in the lung [50].
Although chemokine-GPCR (G-protein-coupled receptor) signaling is known to mediate integrin activation and cytoskeletal remodeling of neutrophils during inflammation [52], a role of GPCRs in neutrophil margination within the noninflamed lung is unclear. Recently, Devi et al. [57] provided evidence to support a role for chemokine signaling in neutrophil margination in the noninflamed lung. Using both mice and primates, Devi et al. [57] demonstrated that neutrophils marginated within the lung show a relatively higher expression of the chemokine receptor CXCR4, which facilitates neutrophil margination in the healthy lung by binding to its ligand CXCL-12 (SDF-1) expressed on the pulmonary capillary endothelium. Taken together, these studies suggest that the role of adhesion in neutrophil margination in the healthy lung cannot be excluded but it seems to be less important than the role of neutrophil resistance to deformation and the presence of the large number of capillary segments with diameters smaller than the neutrophil.
Leukocyte Kinetics in the Inflamed Lung
Neutrophil recruitment to a site of systemic inflammation takes place primarily in the post-capillary venules through sequential steps of rolling, activation, arrest, trans-luminal crawling, trans-endothelial migration, and migration across the ECM into the inflammatory foci [6]. PSGL-1 on neutrophils binds to members of the selectin family (P-, E-selectin on the endothelium, and L-selectin on neutrophils) to mediate rolling, while β2-integrins (LFA-1 and Mac-1) on neutrophils bind to ICAM-1 on the inflamed endothelium or ECM components to mediate arrest and subsequent steps of migration, respectively [6, 58]. Unlike the systemic microcirculation, neutrophil recruitment to the inflamed lungs occurs predominantly (>90 %) in the pulmonary capillary network with minimal contribution (<10 %) from the pulmonary arterioles and venules [2, 4, 45, 46].
In vivo studies performed in dogs , mice, rabbits, and other rodents using diverse pulmonary or systemic stimuli have been instrumental in establishing the current paradigm of neutrophil recruitment in the inflamed lung. Intratracheal (IT) [13, 59, 60], intraperitoneal (IP) [61] and intravascular (IV) administration [62, 63] of gram-negative bacterial endotoxin, lipopolysaccharide (LPS), has been shown to induce neutrophil sequestration in the pulmonary capillaries of mice. Neutrophils have also been shown to sequester in the pulmonary capillaries of the canine [45], mouse [53] and rabbit [64] lung following IV infusion of complement fragments. In addition to these studies, hyper-mechanical ventilation [65], IV IL-6 [66], IV cobra venom factor (CVF) [50, 67], IV activated plasma [68], IT IgG immune complex [67, 69], IT IL-1β [70], IT phorbol-12-myristate-13-acetate (PMA) [56] and IT hydrochloric acid (HCl) [71] have been used to induce pulmonary inflammation in animal models. Also IT instillation of pneumonia inducing microbes such as Escherichia coli (E. coli) [53, 72, 73], Streptococcus pneumoniae (S. pneumoniae) [49, 53, 56, 72], Pseudomonas aeruginosa (P. aeruginosa) [74, 75] and Staphylococcus aureus (S. aureus) [73] have been widely used in animal models to elucidate the molecular and biophysical mechanism of neutrophil recruitment in the inflamed lung.
The release of pathogen or danger associated molecular patterns (PAMPs or DAMPs) by microbial pathogens or necrotic host cells, respectively, is central to the progression of inflammation [76]. The recognition of PAMPs and DAMPs by the pattern recognition receptors (PPRs) on alveolar macrophages, pulmonary endothelial cells, alveolar epithelial cells, and interstitial mast cells results in the release of pro-inflammatory mediators such as IL-1β, TNF, IL-6, G-CSF, and chemokines such as IL-8 (CXCL8; human), KC (CXCL1; mice), MIP2α (CXCL2; mice), and MCP-1 (CCL2) [5, 76]. These mediators cause activation of the neutrophils and endothelial cells by binding to their cognate receptors expressed on these cell types. Activated neutrophils sequester within the pulmonary microcirculation and migrate across the endothelial-epithelial barrier into the alveolar air space. Emigrated neutrophils may release chemokines to further amplify neutrophil recruitment and undergo degranulation, oxidative burst, and NETosis [5]. NETosis involves the release of neutrophil extracellular traps (NETs), which are composed of strands of decondensed chromatin decorated with histones and granule proteins such as neutrophil elastase (NE) or myeloperoxidase (MPO). NETosis has been described in detail elsewhere [12, 77]. These effector functions of neutrophils which are critical to the clearance of bacteria and the resolution of inflammation are tightly regulated; otherwise, the release of neutrophil derived oxidants and proteases can contribute to the disruption of the endothelial–epithelial barrier [76, 78, 79]. Eventually, apoptotic neutrophils are cleared away from the alveolar air space by alveolar macrophages through the process of “efferocytosis” [5, 80, 81].
Neutrophil recruitment into the air spaces of the inflamed lung can be roughly classified into five sequential steps (Fig. 2.1): capillary sequestration and retention, trans-luminal crawling, trans-endothelial migration, trans-interstitial migration, and trans-epithelial migration [2, 4, 5, 59, 76, 82]. In the following text, capillary sequestration and retention is described as a single step, while trans-luminal crawling, trans-endothelial migration, trans-interstitial migration, and trans-epithelial migration are discussed collectively under “Neutrophil emigration into the alveolar air space.”
Neutrophil Sequestration and Retention in the Pulmonary Capillaries
In response to a systemic or pulmonary inflammatory insult, neutrophils sequester within the pulmonary capillaries in large numbers which exceed the baseline marginated pool by several folds [4]. In fact, acute neutrophil sequestration in the inflamed lung is known to be associated with transient peripheral blood neutropenia [45, 64, 72]. Entrapment in the pulmonary capillaries (step 1 in Fig. 2.1) is one of the primary steps in the recruitment of neutrophils to the alveolar air spaces during pulmonary inflammation [2, 4]. Unlike the well-characterized multistep adhesion cascade that mediates neutrophil-endothelium interaction in the inflamed venules [58]; the molecular and biophysical mechanism that enables neutrophil entrapment in the inflamed pulmonary capillaries is incompletely understood [2, 76].
As described in the section “Leukocyte kinetics in the noninflamed lung ,” neutrophils pause in both the straight segments and junctions of the pulmonary capillaries in the noninflamed lung. Neutrophils are much smaller in diameter than most of the pulmonary capillaries and have to deform into an elliptical shape prior to entering a pulmonary capillary segment. Once inside the capillary, the forces of deformation push the neutrophil membrane against the capillary wall, resulting in a large surface area of contact with the capillary endothelium [7, 43]. Neutrophils constitutively express PSGL-1 [83, 84], L-selectin, inactivated β2-integrins, and other glycoprotein ligands for E-selectin [52]. ICAM-1 is known to be constitutively expressed on the lung capillary endothelium and the expression increases following pulmonary inflammation [46, 85]. Based on the quintessential role of these adhesion molecules in the systemic microcirculation and the large contact area that exists between an elliptical neutrophil and the pulmonary capillary endothelium, it would be plausible to expect a role for adhesion molecules in neutrophil sequestration in the inflamed pulmonary capillaries.
The role for selectins in mediating neutrophil capillary sequestration during pulmonary inflammation was supported by early studies. Selectin-IgG chimeras were used to show that P- and L-selectin mediate IV cobra venom factor (CVF)-induced lung injury in rats, while L-selectin and E-selectin mediate IT IgG-immune complex induced lung injury in rats [86] and a P-selectin like epitope is upregulated on the lung capillary endothelium of rats following CVF infusion [87]. Also Mulligan et al. [67] demonstrated that the treatment of rats with an L-selectin blocking Ab inhibited neutrophil recruitment in the lungs following IV infusion of CVF or IT instillation of an IgG-immune complex. However, Bullard et al. [49] raised skepticism about the role of both P-selectin and ICAM-1 by showing that neutrophil recruitment during S. pneumoniae induced pneumonia was unchanged in the lungs of P-selectin/ICAM-1 double mutant mice compared to wild type mice, suggesting no role for P-selectin or ICAM-1.
The discrepancy in these early reports was explained by Doerschuk et al. [50] using a mouse model of CVF-induced lung injury . In this study [50], Abs against P-selectin and ICAM-1 inhibited neutrophil sequestration in the pulmonary capillaries supporting a role for endothelial P-selectin and ICAM-1 [50]; however, mutant mice lacking P-selectin, ICAM-1 or both had no defect in CVF induced neutrophil sequestration, suggesting that neither P-selectin nor ICAM-1 is required [50]. Similarly, functional blocking Abs against ICAM-1 and β2-integrins, LFA-1, and Mac-1, were shown to partially inhibit the acute neutrophil recruitment in the lungs of mice challenged with IT endotoxin [88] or P. aeruginosa [75]; however, the recruitment was completely normal in ICAM-1 deficient mice challenged with either stimuli [75, 88]. As an attempt to explain the difference in the role for adhesion molecules based on functional blocking vs. genetic deletion, it has been suggested that the immune system in mutant animals may have evolved at the embryonic stage to substitute the roles of these molecules with some other molecules or the Abs may inhibit neutrophil adhesion by other mechanisms in addition to functional blocking [4, 49, 50].
Understanding the role of adhesion molecules in neutrophil sequestration in the pulmonary capillaries was further refined by later studies aimed at the temporal kinetics of neutrophil dynamics in the inflamed lung. Using IV infusion of complement fragments in L-selectin deficient mice, Doyle et al. [53] revealed that L-selectin is not required for the initial rapid (<1 min) sequestration but is necessary for the prolonged retention (>5 min) of the sequestered neutrophils within the pulmonary capillaries. In vivo studies using functional blocking monoclonal Abs revealed that neither β2-integrins (CD18) nor L- or P-selectin was essential for the initial sequestration of neutrophils in the pulmonary capillaries of rabbits immediately following infusion of complement fragments [64] or activated plasma [68]. However, both β2-integrins and L-selectin were required to keep the neutrophils arrested within the pulmonary capillaries for greater than 4–7 min [64, 68]. Similarly, Kuebler et al. [63] demonstrated that L-selectin was required for prolonged leukocyte sequestration in the pulmonary capillaries of rabbits following IV infusion of LPS. Also, ICAM-1 was shown to be upregulated on the rat lung capillary endothelium following LPS treatment and mediate prolonged leukocyte sequestration by binding to β2-integrins expressed on stimulated leukocytes [46, 89].
Further studies explored the hypothesis that neutrophil-endothelium interactions in the inflamed lung can be stimulus dependent. Doyle et al. [53] have shown that L-selectin plays a role in neutrophil sequestration within the pulmonary capillaries during E. coli but not S. pneumoniae induced pneumonia, suggesting that the requirement for L-selectin could be possibly dictated by the type of stimulus. Similarly, Doerschuk et al. [56] reported an important observation that β2-integrins are required for prolonged pulmonary capillary sequestration and the recruitment of neutrophils in response to an IT challenge of rabbits with PMA but not S. pneumoniae or HCl, suggesting that the requirement of β2-integrins might also be dependent on the stimuli inducing the pulmonary inflammatory response [56]. This observation was further supported by animal studies performed with diverse inflammatory stimuli. β2-integrin-ICAM-1 adhesion was shown to mediate neutrophil sequestration in the pulmonary capillaries following IV infusion of CVF [90], intrapulmonary instillation of IgG immune complex [69] in rats and intrabronchial challenge with IL-1α but not complement fragments in rabbits [70]. Using a functional blocking Ab against CD18, neutrophil recruitment in the rabbit lungs was shown to be CD18-dependent following IT administration of P. aeruginosa [74] and intrapulmonary E. coli [73] but CD18-independent in response to intrapulmonary S. aureus [73]. Also, acute neutrophil sequestration in the pulmonary capillaries of rats during HCl induced pneumonia was shown to be independent of β2-integrin Mac-1 [91]. Using functional blocking Abs and mice deficient in LFA-1 or ICAM-1, LFA-1-ICAM-1 interaction was shown to be responsible for 50 % of neutrophil recruitment in the lung in response to aerosolized LPS [82]. As shown by the previous studies, the remaining 50 % of recruitment was believed to be mediated by CD18-independent mechanisms [82]. Thus, the requirement of both β2-integrins and L-selectin for prolonged (>1 min) neutrophil sequestration within the inflamed pulmonary capillaries is dictated by the stimulus eliciting the pulmonary inflammatory response [4].
Since adhesion molecules do not seem to be required for the initial (<1 min) sequestration of neutrophils, it is believed that pulmonary inflammation leads to stiffening of neutrophils and a reduction in their deformability, which may result in slower transit and acute mechanical sequestration of neutrophils within the pulmonary capillaries [4]. Cytoskeletal remodeling in neutrophils occurs following the interaction of neutrophil receptors with the inflammatory mediators, cytokines, and danger signals released by tissue macrophages, necrotic host cells, mast cells, and endothelial cells in response to a pulmonary or systemic insult [3, 5, 6, 58]. Several in vitro and in vivo studies support the role of mechanical stiffening in the initial (within <1 min) sequestration of neutrophils within the inflamed pulmonary capillaries. Suwa et al. [66] has shown that the high F-actin content in circulating neutrophils was associated with reduced deformability and an increased retention in the pulmonary capillaries of rabbits. Also, IV infusion of complement fragments in rabbits was shown to cause rapid sequestration of neutrophils in the pulmonary capillaries [92]. Remarkably, the immuno-electron microscopy of the rabbit lung tissue revealed that the sequestered neutrophils were less deformable, round but not elongated in shape and had an actin rich layer under the plasma membrane [92]. Choudhury et al. [65] demonstrated that the initial neutrophil sequestration within the pulmonary capillaries of mice challenged with injurious mechanical ventilation was mediated by stiffening of neutrophils, while the prolonged retention was dependent on L-selectin but not β2-integrins. Also using a rat model of bacterial pneumonia, Yoshida et al. [72] was able to show that the inflammatory mediators in the blood induced cytoskeletal changes resulting in the formation of an F-actin rim under the plasma membrane of circulating neutrophils. These F-actin-rim containing neutrophils were found to be preferentially sequestering within the lungs, which was independent of the β2-integrin-mediated adhesion to the vascular wall [72]. These findings suggested that inflammatory mediators induce a reduction in neutrophil deformability that is responsible for their initial (<1 min) sequestration during pulmonary inflammation.
Based upon the collective evidence, the acute neutrophil entrapment in the inflamed pulmonary capillaries is believed to occur in two steps [4]: The first step involves rapid (<1 min) mechanical sequestration of stiff neutrophils within the pulmonary capillaries. Mechanical sequestration is immediately followed by the second step of prolonged retention (>5 min) that is mediated by adhesive interactions between the sequestered neutrophils and the inflamed capillary endothelium. Depending upon the inflammatory stimulus, the neutrophil-endothelium adhesion during prolonged retention can be either mediated by β2-integrins and L-selectin on neutrophils binding to their cognate ligands on the inflamed endothelium or adhesive pathways that are still elusive.
Although P-selectin is critical for neutrophil rolling along the inflamed systemic venules [93], the role for P-selectin in neutrophil sequestration in the pulmonary capillaries has been treated with skepticism because the immunohistochemistry studies to date were unable to detect P-selectin expression on the lung capillary endothelium [2, 4, 11]. However, Weibel-Palade bodies (which in venules are the vehicles for rapid P-selectin expression on the endothelium) have been shown to be present in the lung endothelium [2, 94]. In a recent study, P-selectin expression was observed on the lung endothelium of mice, which was upregulated 6 h following LPS infusion [95]; however, although 6 h is enough for relocation of P-selectin to the endothelial membrane, the likelihood of P-selectin expressed on platelets adhering to the endothelium was not ruled out [95]. Another study on the role of P-selectin in acid-induced lung injury revealed that intravascular sequestration of neutrophils in the lungs is reduced in mice with no endothelial P-selectin compared to wild type mice, although the lack of P-selectin did not lead to a reduction in lung injury [71]. Asaduzzaman et al. [96, 97] and Roller et al. [47] have also shown that both endothelial and platelet P-selectin contribute to neutrophil sequestration in the lung microcirculation during abdominal sepsis-driven ALI by binding to neutrophil PSGL-1. Recently, a recombinant form of PSGL-1-Ig fusion protein attenuated neutrophil recruitment in the lungs of endotoxin challenged mice, suggesting a role for P-selectin–PSGL-1 interaction in neutrophil sequestration [98]. In addition to P-selectin, E-selectin can also bind to neutrophil PSGL-1 and may mediate neutrophil sequestration; however, E-selectin is not expressed on the noninflamed pulmonary capillary endothelium [4]. Also unlike P-selectin, which is stored preformed in Weibel-Palade bodies of endothelial cells and expressed on the cell surface within minutes in response to an inflammatory stimulus [99], E-selectin requires de novo mRNA synthesis which is unlikely to happen during the acute (1–5 min) neutrophil sequestration response following an inflammatory challenge.
Besides L-selectin and β2-integrins, recent studies have shown that the endothelial surface layer (ESL) and platelets can play a role in neutrophil–endothelium interactions in the inflamed lung. Degradation of the ESL by endothelial heparanase has been suggested to mediate neutrophil sequestration within the pulmonary microcirculation of mice following systemic LPS challenge [20]. Degradation of the ESL was shown to expose ICAM-1 on the pulmonary endothelium, which is normally hidden within the thick ESL and is thus inaccessible to leukocyte β2-integrins [20]. There is also growing evidence to support a role for platelets in neutrophil sequestration and recruitment within inflamed lungs. Neutrophil sequestration in the pulmonary capillaries of mice has been shown to be mediated by platelet-neutrophil aggregation during HCl [71] or endotoxin [78] induced pneumonia and transfusion [100] or abdominal sepsis [47, 96] induced ALI. This platelet-neutrophil aggregation was shown to be mediated by P-selectin on activated platelets binding to PSGL-1 on neutrophils [71, 78, 100] and the PSGL-1-P-selectin engagement triggered outside-in signaling leading to the upregulation of Mac-1 on neutrophils [96, 100].
Neutrophil Emigration into the Alveolar Air Space
The current understanding of the molecular mechanism of neutrophil emigration across the inflamed endothelial–epithelial barrier is based on IVM studies done in nonpulmonary vascular beds in mice [58, 101, 102], in vitro transmigration assays [103–105], electron microscopy and serial section reconstruction of rabbit pulmonary capillaries, arterioles, and venules [2, 104, 106]. Similar to the step of neutrophil sequestration within the lumen of inflamed pulmonary capillaries, the pathway for neutrophil emigration (trans-luminal, trans-endothelial, trans-interstitial, and trans-epithelial migration) into the alveolar air spaces could be CD18-dependent or independent based on the stimulus eliciting the inflammatory response [4]. Since the adhesion pathways mediating CD18-independent emigration are largely unknown, only the molecular events driving CD18-dependent emigration will be discussed here.
In preparation for emigrating across the pulmonary capillary endothelium, arrested neutrophils are required to crawl towards the junctions of endothelial cells (step 2 in Fig. 2.1). ICAM-1 and ICAM-2 are expressed on the pulmonary capillary endothelium [46, 85, 107] and are known to bind activated β2-integrins LFA-1 and Mac-1 on neutrophils to mediate trans-luminal crawling in the systemic venules [102, 105]. The process of trans-luminal crawling requires transient and regional changes in the mechanical properties and adhesion molecule distribution of neutrophils and pulmonary endothelial cells [4, 76]. Signaling pathways triggered by CD18-ICAM-1 mediated neutrophil–endothelial adhesion, chemokine–GPCR interaction, and chemokine gradient sensing collectively result in the activation of small GTPases and the actomyosin machinery within the neutrophils [6, 52, 58, 100, 101]. These activation events enable trans-luminal crawling of leukocytes by triggering spatial and temporal changes in the cytoskeletal organization, generation of a protrusive leading edge and a contractile uropod, clustering of PSGL-1 and recycling of CD18 to the leading edge of the leukocytes. Recently, P-selectin-PSGL-1-mediated platelet interactions with adhered neutrophils have also been shown to trigger signaling events that result in neutrophil polarization, redistribution of CD18 and CXCR2 and trans-luminal crawling in the inflamed cremaster venules of mice [100]. Although platelet-neutrophil aggregation is known to occur in the inflamed pulmonary microcirculation [78], it is not known whether these interactions contribute to intra-luminal crawling within the pulmonary capillaries.
The current understanding of the molecular mechanism of leukocyte trans-endothelial migration (TEM; step 3 in Fig. 2.1) across pulmonary capillaries is largely based on in vitro cell culture transmigration studies or IVM studies of the systemic microcirculation in mice. Due to the difference in the primary site of neutrophil recruitment in the lungs (pulmonary capillaries) vs. systemic circulation (post-capillary venules), the molecular mechanism of TEM in the two vascular beds may not be exactly the same. Nevertheless, the current paradigm of TEM serves to explain the kinetics of leukocyte trafficking in the inflamed lungs. Based on in vitro endothelial cell culture studies [103–105] and electron microscopy of the rabbit lung during S. pneumoniae induced pneumonia [2, 106], leukocytes crawling in the pulmonary capillaries are believed to undergo TEM in less than 2 min [104]. TEM occurs predominantly (60 %) through a paracellular (between endothelial cells) pathway at the junction of three endothelial cells (tricellular corners) and less frequently (20 %) at the junction of two endothelial cells (bicellular corners) or (20 %) through the cytoplasm (transcellular pathway) of endothelial cells.
The paracellular TEM of an adhered leukocyte is regulated by endothelial cell junction integrity, which is dependent on the two primary junctional structures “adherens junctions” and “tight junctions” located within the endothelial cleft [2, 5, 52, 58, 104, 105, 108–111]. Adherens junctions are formed by homophilic interactions between VE-cadherin molecules on adjacent endothelial cells [112]. The stability of adherens junctions is dependent on the linkage of VE-cadherin to the endothelial cell actin-cytoskeleton by cytoplasmic catenins (α, β, and γ) [58]. Tight junctions consist of homophilic interactions between members of the junctional adhesion molecule (JAM) family (JAM-A, JAM-B, JAM-C, and endothelial cell-selective adhesion molecule (ESAM)) and claudins [112]. Depending on the vascular bed and the inflammatory stimulus, the borders of adjacent endothelial cells can also express PECAM-1, CD99, ICAM-2, and the polio virus receptor (PVR) [58, 108]. Human lung endothelial junctions are known to express JAM-A and C, while mouse lung endothelial junctions express only JAM-A [2]. PECAM-1 and CD99 contribute to the endothelial cell junction integrity by undergoing homophilic interaction [108].
Related posts:
 Sickle Cell Disease and Acute Chest Syndrome: Mechanisms and Pathogenenesis
Sickle Cell Disease and Acute Chest Syndrome: Mechanisms and Pathogenenesis
 Acute Pulmonary Complications of Bone Marrow and Stem Cell Transplantation
Acute Pulmonary Complications of Bone Marrow and Stem Cell Transplantation
 Acute Pulmonary Manifestations of Hematologic Malignancies
Acute Pulmonary Manifestations of Hematologic Malignancies
 Storage Lesion: Evolving Concepts and Controversies
Storage Lesion: Evolving Concepts and Controversies
 Sickle Cell Disease and Acute Chest Syndrome: Epidemiology, Diagnosis, Management, Outcomes
Sickle Cell Disease and Acute Chest Syndrome: Epidemiology, Diagnosis, Management, Outcomes
 Transfusion-Related Immunomodulation (TRIM): From Renal Allograft Survival to Postoperative Mortality in Cardiac Surgery
Transfusion-Related Immunomodulation (TRIM): From Renal Allograft Survival to Postoperative Mortality in Cardiac Surgery
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


