Keywords
Lentiviral vector, lentivector, cancer vaccine, gene therapy, immunotherapy, cancer
Introduction
The immune system likely evolved to fight off disease caused by infections with foreign pathogens. However, it has been proposed to also be involved in the fight against cancer, a disease usually caused by self-pathogens . In this scenario, tumor cells are recognized by the immune system as the equivalent of foreign pathogens. This recognition is usually mediated through one or more tumor-associated antigens (TAAs) expressed on the cancerous cells. These TAAs may be expressed uniquely on tumor cells, or they may be expressed in a mutated form or at higher levels in tumor cells compared to normal cells . Such differential expression may allow the immune system to preferentially target harmful tumor cells while ignoring any normal cells that express the TAA. Many different TAAs have been described (reviewed by Cheever et al . ), most from melanomas, which can be more immunogenic than other cancers.
The identification of TAAs opens up the opportunity for therapies that specifically target tumor cells while avoiding healthy normal cells. Among immunotherapies that utilize this approach, monoclonal antibodies have been successful against a variety of different antigens for several different types of cancer. U.S. Food and Drug Administration (FDA)-approved antibodies include Herceptin , rituximab , cetuximab , and bevacizumab , which have been used to treat cancer of the breast, colon, lung, and lymphoma . At the same time, much effort has been put into developing cancer-targeted vaccines . However, the success rate for vaccines has been much lower than that for therapeutic antibodies . Nevertheless, vaccination therapies remain one of the most sought-after treatments because of their potential simplicity and efficacy. The first FDA-approved cancer vaccine involved the delivery of a TAA directly to dendritic cells (DCs) ex vivo , and vaccine protocols utilizing this delivery method were generally more successful than alternative approaches in recent clinical trials . DCs are considered to be the most powerful antigen-presenting cell , and thus a strong immune response may depend on antigen presentation by these cells.
There are several different ways to deliver an antigen to DCs and other antigen-presenting cells (APCs). One of the most commonly used methods involves viral vectors. These have been used to deliver recombinant antigens ever since the first report using vaccinia virus to express the hepatitis B surface antigen . Many different viruses are used as vectors for delivering antigens. The ideal vector would generate a strong immune response to the antigen of interest while generating a minimal response to the viral components of the vector, all while being safe for use in humans . Commonly used viral vectors include adenovirus, vaccinia virus, γ-retroviruses, and lentiviruses, which is the subject of this chapter.
Lentiviral vectors (lentivectors or LVs) have the advantages of efficient transduction of both dividing and nondividing cells , the ability to achieve long-term expression due to integration into the host genome, low natural antivector immunity in most human populations , and a low potential for genotoxicity from insertional mutagenesis . Furthermore, they can carry large genes, allowing for the expression of not only large protein antigens but also target antigens fused to accessory genes that can tailor the immune response. These advantages contrast with other viral vector systems based on adenovirus, subject to antivector immunity; adeno-associated virus, with limited carrying capacity; and γ-retrovirus, with a higher potential for genotoxicity .
LV vaccines are also subject to significant limitations. Because the most advanced LV systems are derived from HIV, a dangerous human pathogen, preventing the development of pathogenic HIV in immunized individuals is an essential safety concern. Perhaps more concerning is the growing evidence that robust immune responses by themselves are not necessarily sufficient to generate any measurable antitumor response . Therefore, a successful vaccine must be able to recruit effector T cells to the tumor environment and counter any immunosuppressive factors present there . To date, there is not enough evidence to determine whether LVs are able to do so, but work with other types of vaccines has uncovered new strategies for overcoming these barriers.
In this chapter, we focus on the origin and history of lentiviral vectors, their use in the transduction of dendritic cells for antigen presentation, successful applications of LV-mediated antigen delivery ex vivo and in vivo , and current and future strategies for clinical trials with LVs.
Structure and Function of Lentiviruses
Classification
Lentiviruses are a genus of viruses within the Orthoretrovirinae subfamily of the Retroviridae family. Members of this genus include pathogens of bovine, equine, feline, ovine, and primate origin . The most prominent member of the genus is the human pathogen, HIV, which infects more than 30 million people worldwide. Like other retroviruses, lentiviruses are enveloped particles that bud from an infected cell’s plasma membrane. These particles are 80–120 nm in diameter, and they contain a single-stranded 9.2-kb RNA genome and several structural proteins, including the matrix, capsid, nucleocapsid, envelope, and reverse transcriptase proteins.
Life Cycle and Gene Function
Lentiviral particles bind to their target cell through an interaction between the cell’s receptor and the virus’s envelope glycoprotein. After fusion of the viral and cellular membranes, the virus core is released into the cytoplasm, where the viral reverse transcriptase reverse transcribes the viral genome into a double-stranded (ds) DNA copy with long terminal repeats (LTRs) at both ends. The viral DNA is transported into the cell nucleus, where it integrates into the host genome and is actively transcribed. Viral transcripts are translated into structural and nonstructural proteins, all of which bud out from the cell membrane to form an immature viral particle. Following budding, the viral proteases cleave the viral proteins into the final products to generate an infectious particle. In a simple orthoretrovirus, such as murine leukemia virus (MLV), the virus proceeds through this life cycle with a genome that encodes three major proteins: gag, pol, and env ( Figure 24.1 ). Lentiviruses encode additional components, such as Tat, Rev/RRE, Vif, Vpr, Vpu, and Nef. The functions of each of these proteins are briefly described in Table 24.1 . Many of these elements are dispensable, and extensive research testing the removal of individual components has led to the development of highly modified LVs. For example, gag, pol, and env can be delivered in trans , but the LTR, integration sequence, packaging site, and transport RNA-binding site must be included in cis .
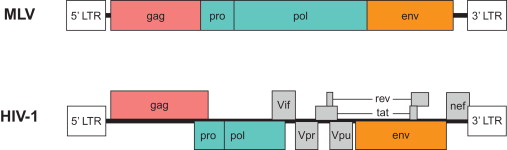
| Protein | Function |
|---|---|
| Gag | Mature protein products MA (matrix), CA (capsid), and NC (nucleocapsid) provide structural support for the viral particle. |
| Pro-Pol | The protease Pro cleaves the viral polyprotein. The RT (reverse transcriptase) polymerizes DNA or RNA templates into dsDNA. The IN (integrase) inserts the viral dsDNA into the host genome. |
| Env | Glycoproteins bind to and trigger fusion with host cell membrane receptors. |
| Accessory proteins | Tat activates transcription. Rev facilitates transport of viral mRNAs to the cytoplasm. Vif promotes viral infectivity. Vpr is a structural component. Vpu enhances viral particle release. Nef is a multifunctional structural protein. |
Lentiviral Vector Systems
Retroviruses were first adapted for use as a gene delivery vector as early as 1983 . Oncoretroviral vector systems have been developed and used for therapies; however, they commonly suffer from problems of particle instability, low titers, and an inability to transduce nondividing cells . To address these problems, HIV and other lentiviruses were adapted for gene delivery . These lentiviral vectors have been developed over the years since then to create safer, more efficient, and more convenient systems that are commonly used today. No one lentivector has proven to be the ideal choice in all situations, but numerous modifications have been made to improve an LV’s suitability to a given situation. This section surveys the different ways LVs have been adapted for different purposes.
Production of Virus
The most common method for producing viral particles is through transient transfection of HEK-293T cells . Following the methods established for oncoretroviruses, the LV components were separated onto three plasmids: a packaging plasmid, a transfer plasmid, and an envelope plasmid ( Figure 24.2 ). These were further refined into the so-called “second-generation” system, in which all viral virulence factors that were unnecessary for gene delivery and expression were removed . Thus, accessory proteins Vpr, Vif, Nef, and Vpu are not present in LVs. The third-generation version separated the Rev gene onto a different plasmid and replaced Tat and the 5′ LTR by a constitutive promoter, usually that of cytomegalovirus (CMV) . With these enhancements, LVs are now less pathogenic, less oncogenic, and more efficient than either their HIV forebears or their oncoretrovirus cousins.
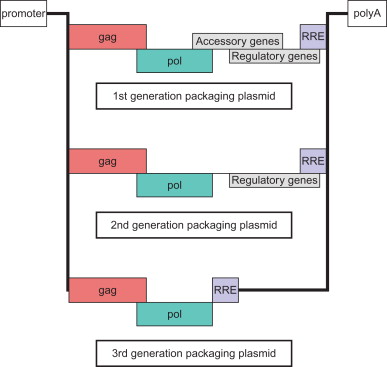
This third-generation production system forms the basis for most of the gene therapy experiments conducted to date. Meanwhile, many groups continue to modify the LV structure in order to improve safety and efficiency. Some of the additional modifications include the following:
- •
Deletion of the U3 region of the 3′ LTR to generate self-inactivating transfer vectors (SINs)
- •
Inclusion of a triple-helix signal (TRIP) for improved nuclear import
- •
Addition of woodchuck hepatitis B post-transcriptional regulatory element (WPRE) to improve gene expression
These modifications have enabled the study of LVs in a diverse array of cell types and tissues. Moreover, the safety improvements are likely to facilitate the transition of LVs into human studies, of which there are currently very few reports.
Altering Viral Tropism
One of the most common variations among LVs in use today is the vector’s tropism, or specificity for different types of host cells or tissue. Tropism can be influenced by both the envelope proteins on the viral particle and the promoter that drives transgene expression. The former enhances specificity by driving infection toward certain cell types. The latter enhances specificity by limiting active transgene transcription to certain cell types. By modifying either of these two components, one can alter the LVs tropism to create LVs with either a very narrow or a very broad host cell range. Both approaches can be effective at directing the transgene expression to specific compartments and are important for generating antitumor immunity.
Lentiviral vectors are flexible in the glycoproteins they incorporate into their viral envelope. HIV’s native envelope protein, gp120, has a limited host range and confers relatively poor stability to viral particles. Therefore, virtually all LVs are pseudotyped by incorporating an envelope protein from another virus into the LV particle. Many other glycoproteins have been used for pseudotyping, each with its own advantages and disadvantages ( Table 24.2 ). The glycoprotein from vesicular stomatitis protein (VSVg) is most commonly used for LVs because it infects a broad range of cell types and helps to produce more stable viral particles . On the other hand, this protein can be toxic when constitutively expressed , which may limit its use in large-scale LV production. For vaccine therapies, VSVg has been successful in infecting APCs, although other envelope proteins, such as sindbis virus glycoprotein (SVg), have been engineered for greater specificity and efficacy .
| Family | Genus | Vector | Target | References |
|---|---|---|---|---|
| Rhabdoviridae |
| HIV-1, SIV HIV-1 | Broad host cell range Neurons | |
| Arenaviridae | Arenavirus Lymphocytic choriomeningitic virus | HIV-1, SIV | Cancer cells and astrocytes | |
| Flaviviridae | Hepacivirus Hepatitis C virus | HIV-1 | CD81 + cells, including hepatoma cells | |
| Paramixoviridae | Respirovirus Sendai virus | HIV-1, SIV | Hepatocytes | |
| Baculoviridae | Nucleopolyhedrovirus Fowl plague virus | HIV-1, SIV | Broad host cell range with poor transduction of hematopoietic cells | |
| Filoviridae |
| HIV-1 | Lung cells and myocytes | |
| Retroviridae | Alpharetrovirus, Jaagsiekte sheep, Ross River, Semliki Forest virus, Sindbis virus | HIV-1 | Lung cells | |
| HIV-1, SIV | Cancer cells |
Tissue-specific promoters have been successfully used to direct expression of LV-encoded genes to those cell types in which they are best used. The promoters of greatest interest to the gene therapy of cancer include the α-fetoprotein liver-specific promoter for hepatoma targeting , and the prostate specific antigen promoter for prostate cancer targeting . LVs utilizing these promoters have been engineered to deliver toxins or suicide signals to the cancerous cells as a strategy to eliminate cancer. In addition, promoters such as the HLA-DRα major histocompatibility complex (MHC) class II-specific promoter have been tested for directing transgene expression to APCs such as DCs . LVs using this type of promoter may be able to generate a stronger antitumor immune response by efficiently expressing and presenting TAAs in APCs while minimizing expression and presentation in normal healthy cells.
Another modification to the LV transgene promoter that may be applicable to cancer therapy is the use of inducible promoters. Most commonly studied is the tet-on system, in which transgene expression is repressed in the absence of tetracycline . In these experiments, the authors found that gene expression could be tightly and rapidly regulated by administration of tetracycline. This promoter modification offers the possibility of turning on gene expression only for a limited time by administering tetracycline to a patient. By minimizing the duration of expression of the transgene, the immune response to an antigen might be better regulated, and the potential for adverse side effects caused by transgene expression may be reduced.
Integration-Deficient Lentivectors
Another modification to the LV structure that may improve the safety of these vectors is the development of integration-deficient LVs (IDLVs). These vectors came about with the discovery that retroviruses do not always need to integrate their genomes into the host genome in order to transcribe their genes. When a lentivirus such as HIV infects a cell, not only does it integrate its genome into the host but also it produces circular episomal DNA of its genome. These episomes have been shown to be transcriptionally active, albeit at a much lower rate . Nevertheless, in many circumstances, this level of expression is sufficient to generate enough antigens to prime an effective immune response. Thus, if one is able to eliminate the integration process, LVs can transduce cells without the safety concerns involved with insertional mutagenesis.
Toward this goal, several mutations in either the integrase protein or the att DNA attachment site on U3 have been found to impair integration . With these modifications, and with the introduction of SIN vectors, which improves episomal gene expression , IDLVs encoding various antigens have been shown to lead to a CD8 + T cell response and in some cases antitumor immunity . Notably, using IDLV generally requires administering a higher dose than that required for integrating vectors, but the difference is not large enough to prevent their use, given the safety advantages .
As described in the previous two sections, many developments in LV technology, such as SIN LVs, tissue-specific promoters, inducible promoters, envelope modifications, and genetic insulators, may all contribute to reducing the risk of insertional mutagenesis. However, the most effective solution to addressing this safety concern is to use IDLVs because they eliminate the integration step in the LV life cycle. Adding these additional layers of safety precautions to an IDLV should dramatically decrease the likelihood for oncogenesis, replication-competent lentivirus (RCL), or other undesirable side effects.
DC-Targeted Lentivectors
As mentioned previously, pseudotyping and tissue-specific promoters have been used to target LVs to specific tissue types. One particular tissue that is an attractive target for a vaccine is immune cells, especially APCs. By targeting these cells, LVs could preferentially deliver a TAA or other genetic cargo directly to those cells that are responsible for initiating an antigen-specific immune response. Many groups have tested promoters found in APCs , including those of dectin 2, MHC class II, and DC-STAMP. These strategies have been successful at achieving high levels of tissue-specific expression in various APCs. In contrast, strategies incorporating macromolecules into the pseudotyping envelope have been often limited by low transduction efficiency . These include attempts to incorporate ligands, peptides, cytokines, or single-chain antibodies. One major exception is the modification of other viral glycoproteins and inclusion of them in the LV production process. For example, Lin et al . found that including a modified MLV envelope protein and a binding-defective influenza hemagglutinin (HA) protein allowed for the efficient binding and fusion of LV particles to cells expressing the MLV target, Flt-3 . Similarly, Funke et al . found that combining a measles virus hemagglutinin (H) with a CD20-binding single-chain antibody could lead to higher and more specific transduction of CD20 + B cells than LVs pseudotyped with the generic VSVg glycoprotein .
This strategy of separating the binding and fusion functions of a viral envelope protein has also been applied to the sindbis virus glycoprotein (SVg) . Point mutations in the glycoprotein gene can eliminate its binding activity to its natural target, heparin sulfate, without significantly impairing its ability to bind its secondary target, DC-SIGN, or its ability to promote virus–cell fusion . DC-SIGN is a C-type lectin expressed on the surface of DCs that is involved in T cell/DC interactions . Thus, LVs pseudotyped with this modified SVg protein can specifically infect and express their cargo in DCs. By targeting a strong APC such as DCs, these vaccines can expedite the process of antigen recognition and may lead to an enhanced adaptive immune response.
Large-Scale Production
Most of the completed research on lentiviral vaccines has been performed in the laboratory or in small clinical trials. For a therapy to become a viable treatment in the clinic, LVs will have to be produced on a much larger scale than has been published to date. Some of the important variables to consider when attempting to achieve this goal include the cell line, media, and culture vessels used for LV production . In theory, the most practical cell line to utilize would be one that stably expresses the LV components, thus eliminating the need for large-scale transfections. Several such cell lines have been reported ; however, they all only give moderately improved yields compared to transient transfection. In fact, several groups have demonstrated safety and feasibility of large-scale transient transfection . These methods may be the first applied to clinical trials until an improved producer cell line is identified. In addition, improvements to methods of concentrating the virus and consistently measuring viral titer are being pursued . Such advancements will expedite the production of LVs on a large scale.
LV-Mediated Presentation of Antigens
Here, we present a brief overview of the adaptive immune response and its relationship with LV-delivered antigens. For more detail, see Murphy et al . . In order to stimulate a T cell-mediated immune response, antigens must be presented by APCs to T cells. This presentation takes place in the context of MHC proteins, which are divided into two classes. MHC class I present antigens that are derived from the intracellular space, whereas MHC class II present antigens that are derived from the extracellular space. This MHC-mediated presentation of the antigen (or, specifically, its epitopes) is termed “signal 1” of the T cell activation process. In order to fully expand and differentiate naive T cells into effector T cells, APCs must also present signals 2 and 3. Signal 2 is commonly delivered by the costimulatory B7 molecules on the surface of the APC, whereas signal 3 is mediated through cytokines secreted by the APC ( Figure 24.3 ).
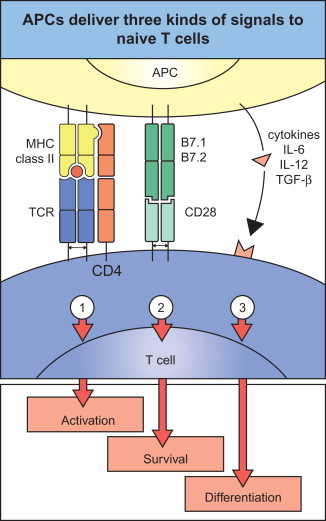
In order to present all three signals, the APC must be properly activated, which usually involves signals delivered by the pathogen or by leukocytes of the innate immune system. In the case of LV vaccines, it appears that the process of LV infection is sufficient to provide all these signals in the infected DCs. Initial studies of LV-transduced DCs found that the transduction of these cells does not impair DC maturation . Further research determined that infection by LVs helps to stimulate the maturation process in some cases. This maturation occurs in the case of a high multiplicity of infection and is TLR3 and TLR7 dependent . Because DCs are the most powerful APC , a successful vaccination protocol will likely induce expression of the target antigen by these cells.
As described previously, antigen presentation can occur through either MHC class I or class II pathways. Each of these pathways leads to activation of a different arm of the adaptive immune system; class I generally directs the response toward an antibody-mediated response, whereas class II directs the response toward a cytotoxic T lymphocyte (CTL) response. For immunity of cancer, most vaccination schemes have sought to stimulate the CTL response to kill the pathogenic tumor cells. Therefore, presentation through the MHC class I response is essential. On the other hand, activation of helper T cells (T H ) through class II presentation has been shown to improve the CTL response . Some studies have reported that LV transduction by itself can effectively activate this CD4 response . Even more elegantly, several groups have shown that fusion of the invariant chain or transferrin receptor to the transgene can artificially direct that antigen toward the MHC class II pathway .
It is likely that no one LV vaccine will be ideal for all situations. Understanding the details of how the immune system responds to a given vaccine may allow us to develop specialized vaccines that are more effective. Furthermore, being able to push the immune system toward a CTL or T H response could be a powerful asset in the LV toolkit.
LV Transduction of DCs Ex Vivo
A broad body of work has established that LVs can be used as vaccines by directly transducing DCs that have been harvested ex vivo and then reintroducing the DCs back into the patient for antigen presentation ( Table 24.3 ). These experiments form the basis for future direct immunizations with LVs (discussed later) and have proceeded into clinical trials.
| Tumor Antigen | Vector Type | Vaccine Details | Immunity | Antitumor Response | References |
|---|---|---|---|---|---|
| Hepatoma mTAAs (Sca-2, GP38, RABP1) |
| Twice s.c. injection of LV-transduced BMDCs in BALB/c every week | CTL, secreting IFN-γ | Therapeutic against hepatoma cell line | |
| Prostate cancer murine erbB2 transgenic |
| Twice i.p. injection of LV-transduced BMDCs in C57BL/6 every 2 weeks | erbB2tr-specific Th1 cytokine and antibody | Protective | |
| Melanoma human MAGE-A3 |
| Human MoDCs transduced by LV | CTL (new epitope EGDCAPEEK) | N.D. | |
| Melanoma murine TRP2 |
| s.c. injection of LV-transduced BMDCs in C57BL/6 | T cell proliferation | Protective and therapeutic | |
| Melanoma human Melan-A |
| Human MoDCs transduced by LV | Antigen-specific CD8 T cells, secreting IFN-γ | N.D. | |
| Melanoma human MART1, murine TRP2 | HIV-1 SIN LV/3 VSV-G | s.c. injection of BM cells cotransduced by LV-GM-CSF/IL-4 + LV-hMART1 and mTRP2 in C57BL/6 | Antigen-specific CD8 T cells; CTL, secreting IFN-γ | Protective | |
| Melanoma human tyrosinase |
| Human APCs transduced by LV | Activation of Ag-specific TIL | N.D. |
a Listed are experiments that utilized LVs to transduce APCs harvested from humans or mice, grouped by the target antigen.
LVs Can Transduce DCs In Vitro
One of the initial limitations of using ex vivo harvested DCs in an immunization regimen was the fact that the DCs harvested were quiescent progenitor cells . Thus, LVs’ ability to transduce nondividing cells ended up being an important advantage to this type of procedure. The first report of LV-transduced DCs was by Unutmaz et al ., who used human monocyte-derived DCs . Since then, numerous groups have described the successful transduction of monocyte-derived and CD34 + -derived human DCs , as well as mouse DCs . Each of these groups was able to show varying efficiency of expression, and in many cases transgene expression persisted indefinitely.
LV-Transduced DCs Can Present Antigens
Knowing that LVs could be used to transduce DCs, the next question to be asked was whether these transduced DCs were able to process antigens properly and present them to T cells. To answer this question, harvested DCs were transduced with various antigens and incubated with established T cell clones in vitro . If the DCs were able to process antigens and present them to T cells properly, these T cells would receive activation signals and mature into activated T cells. Such activation was observed in response to transduction with the laboratory antigen OVA , as well as TAAs MAGE-3 , Melan-A/MART-1 , TRP-2 , and tyrosinase . Thus, it appears that infection of DCs by LVs does not hinder these cells’ ability to undergo the normal process of antigen presentation.
LV-Transduced DCs Can Activate T Cells In Vitro
Not only could these transduced DCs activate T cell clones but also they could activate naive harvested T cells in vitro . Firat et al . found that CD8 T cells could be activated by MART-1-transduced DCs . Breckpot et al . demonstrated priming of CD8 T cells and CD4 T cells when cultured with DCs transduced with a MAGE-A3 antigen fused to the invariant chain . These results implied that these DCs could activate both major branches of T cells in vitro , and the next step was to test their efficacy in vivo .
LV-Transduced DCs Can Protect against Tumor Challenge
To test the in vivo efficacy of LV-transduced DC presentation, derived DCs were transduced with various antigens and then injected back into mice. Both the immune response and an antitumor response were measured. Dullaers et al . found that immunization with these DCs led to a strong CD8 T cell response to the OVA antigen, and they were able to demonstrate specific lysis of OVA-expressing tumor cells in vitro . Furthermore, they showed that mice immunized with these DCs were protected against tumor challenge. Therapeutic immunization did not cure preexisting tumors but did slow their growth.
These results might not be the best model for human cancers because OVA is not a self-antigen. However, Metharom et al . observed similar results when they tested the protection of DC immunization against tumors expressing the self-antigen TRP-2 . Again, therapeutic immunization did not cure preexisting tumors, but it slowed their growth. In total, these experiments showed that LV transduction of harvested DCs can elicit a strong immune response to numerous antigens, and this response is sufficient to provide protection against tumor growth.
Currently, ex vivo transduction of DCs has progressed the furthest toward a viable therapy. Many clinical trials have been performed with DCs that were loaded ex vivo by means other than LV transduction. Thus, there is a large base of knowledge on which to adapt the LV transduction method. However, ex vivo loading of DCs is limited by the cost and difficulty of harvesting DCs from each individual patient, and a vaccination method that could bypass this step would represent a major improvement.
LV Immunization in Vivo
Promising results from ex vivo DC transduction experiments led to the adaptation of such an approach for in vivo direct injection of LV vectors. As with any in vivo adaptation, much of the mechanisms by which these LV vectors work have yet to be fully uncovered. However, initial results in preclinical models have been promising, and numerous modifications have been discovered that can improve the immune response for these vectors. These experiments are summarized in Table 24.4 .
| Tumor Antigen | Vector Type | Vaccine Details | Immunity Observed | Antitumor Response | References |
|---|---|---|---|---|---|
| Melanoma polyepitope | HIV-1 LV/1 VSV-G | s.c., i.v., or i.p. injection of LV in HHD mice | Vigorous and multiply-specific long-term CTL | N.D. | |
| Melanoma human NY-ESO-1 | HIV-1 SIN LV/3 VSV-G | s.c. injection of LV in HLA-A2/H-2K b mice | Ag-specific T and B cell responses | N.D. | |
| Melanoma human NY-ESO-1 |
| s.c. or i.v. injection of LV in HHD mice; or boost with VV-NY-ESO-1 | Ag-specific CD4 and CD8 T cells; CTL, boost improves response | N.D. | |
| Melanoma human Melan-A/ELA 26–35 | HIV-1 SIN LV/3 VSV-G | s.c. injection of LV in HLA-A2/H-2K b mice; or boost with the same LV | Ag-specific CD8 T cells | N.D. | |
| Melanoma human Melan-A 26–35 | HIV-1 SIN LV/3 VSV-G | s.c. injection of LV in HLA-A2/H-2K b mice; or IL-7 as adjuvant | Ag-specific CD8 memory T cells; CTL | N.D. | |
| Melanoma murine mutated and wt TRP1 | HIV-1 SIN LV/3 VSV-G | Footpad injection of LV in C57BL/6 | CD8 T cells recognizing multiepitopes; mutant > wt; CTL cytokines; TIL | Protective and therapeutic | |
| Human telomerase reverse transcriptase | HIV-1 SIN LV/2 VSV-G | s.c. injection of LV in HHD mice; or boost with peptide/IFA | Potent and diversified CTL, secreting IFN-γ | Protective and therapeutic | |
| NY-ESO-1 | Dectin-2 promoter VV-boost | i.v. C57 BL/6 mice | CTL, epitope-specific CD4 T cell | N.D. | |
| HIV-1 SIN LV/2 VSV-G | s.c. injection of LV in C57BL/6 or BALB/c-Neu, rat Her-2/neu transgenic | Ag-specific CD8 T cells, secreting IFN-γ | Therapeutic | |
| Melanoma murine TRP2, CMV, and MHCII promoters | HIV-1 SIN LV/3 VSV-G | Twice i.v. injections of LV in C57BL/6, 1 week apart | CMV: Ag-specific CD8 T cells, secreting IFN-γ MHCII: None | Protective |
Related posts:
 Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
 Genetically Engineered (T Cell Receptor) T Cells for Adoptive Therapy
Genetically Engineered (T Cell Receptor) T Cells for Adoptive Therapy
 Imaging of Oncolytic Virus Gene Expression
Ethics in Translational Gene Transfer Research
Imaging of Oncolytic Virus Gene Expression
Ethics in Translational Gene Transfer Research
 Viral Insertion Site Detection and Analysis in Cancer Gene Therapy
Viral Insertion Site Detection and Analysis in Cancer Gene Therapy
 Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


