This article presents an overview of the current literature about the biology, pathology, and the clinical management of leiomyosarcoma. In addition, the article emphasizes and discusses the current systemic treatment options available for patients with leiomyosarcoma, which range from cytotoxic chemotherapy to target therapies. Particular leiomyosarcoma subtypes, such as uterine leiomyosarcoma and inferior vena cava leiomyosarcoma, are discussed separately.
Key points
- •
Leiomyosarcoma is a malignant mesenchymal tumor that derives from the smooth muscle lineage.
- •
The underlying genetic mechanisms remain unclear, and complex and unbalanced karyotypic defects are the only shared features observed across the different leiomyosarcoma subtypes.
- •
Cell-cycle perturbations, mainly through RB1 defects, and phosphatidylinositol 3 kinase/Akt pathway activation caused by PTEN genomic deletion are the two most consistent drivers commonly observed in sarcomas, including leiomyosarcoma.
- •
Patients with unresectable metastatic leiomyosarcoma are considered incurable and the treatment intention for systemic disease is always palliative.
- •
Unlike other soft tissue sarcomas, leiomyosarcomas are particularly sensitive to the combination of gemcitabine-docetaxel, which is currently regarded as a standard of care in this population.
- •
Chemotherapy treatment with trabectedin has shown exquisite activity in leiomyosarcoma, mainly in the form of long disease stabilization.
- •
Pazopanib is an oral multikinase inhibitor recently approved for the treatment of soft tissue sarcomas in leiomyosarcoma.
Epidemiology
Leiomyosarcoma is one of the most frequent soft tissue sarcomas, with an estimated incidence ranging between 10% and 20% of all newly diagnosed soft tissue sarcomas. Population-based data on the incidence of leiomyosarcoma were investigated in a study from the Surveillance, Epidemiology, and End Results (SEER) program, covering a total of 35,359 soft tissue sarcomas diagnosed during 2005 to 2009. Leiomyosarcoma comprised a significant percentage of soft tissues and abdominal-pelvic sarcomas, only outnumbered by liposarcomas, and is the predominant sarcoma arising from large blood vessels. Aside from these locations, it is less common in the extremities, accounting for perhaps 10% to 15% of limb sarcomas, with a preference for the thigh. In addition, leiomyosarcomas of the uterus, with an estimated incidence of 0.64 cases per 100,000 women, are among the most common uterine sarcomas and likely account for the single largest site-specific group of leiomyosarcomas.
As in soft tissue sarcomas in general, overall incidence of leiomyosarcoma increases with age, and peaks at the seventh decade. By contrast, uterine leiomyosarcoma occurs from the third decade into old age, but is most common in the perimenopausal age group, in the fifth decade. The sex incidence depends on tumor location, with most patients with retroperitoneal and inferior vena cava leiomyosarcoma being women, whereas there is a mild male predominance in noncutaneous soft tissue sites and cutaneous leiomyosarcoma.
Causes and Predisposing Factors
There are few clear causal or predisposing factors identified for this disease. Several exogenous agents have been studied, but only Epstein-Barr virus (EBV) infection, in the setting of severe immunosuppression, has been associated with leiomyosarcomas among patients with acquired immunodeficiency syndrome (AIDS) and after kidney, cardiac, and liver transplantation. Most of EBV-related leiomyosarcomas occur in children and young adults, and develop in organs not traditionally considered preferred sites for leiomyosarcoma. Other traditional risk factors for sarcomas, such as radiotherapy, rarely lead to the development of leiomyosarcomas. In addition, the role for estrogenic stimulation remains unclear. Patients with hereditary retinoblastoma have a cumulative risk of 13.1% for developing any soft tissue sarcoma as a secondary malignancy, including leiomyosarcoma, which further agrees with the relevance of RB1 loss in sporadic leiomyosarcoma (discussed later). In contrast, although clearly reported, leiomyosarcoma is an uncommon subtype of sarcoma among patients with Li-Fraumeni syndrome.
There is no solid evidence showing that leiomyoma (the benign counterpart of leiomyosarcomas) can undergo malignant transformation, and it is currently widely accepted that leiomyosarcomas are tumors that arise de novo.
Epidemiology
Leiomyosarcoma is one of the most frequent soft tissue sarcomas, with an estimated incidence ranging between 10% and 20% of all newly diagnosed soft tissue sarcomas. Population-based data on the incidence of leiomyosarcoma were investigated in a study from the Surveillance, Epidemiology, and End Results (SEER) program, covering a total of 35,359 soft tissue sarcomas diagnosed during 2005 to 2009. Leiomyosarcoma comprised a significant percentage of soft tissues and abdominal-pelvic sarcomas, only outnumbered by liposarcomas, and is the predominant sarcoma arising from large blood vessels. Aside from these locations, it is less common in the extremities, accounting for perhaps 10% to 15% of limb sarcomas, with a preference for the thigh. In addition, leiomyosarcomas of the uterus, with an estimated incidence of 0.64 cases per 100,000 women, are among the most common uterine sarcomas and likely account for the single largest site-specific group of leiomyosarcomas.
As in soft tissue sarcomas in general, overall incidence of leiomyosarcoma increases with age, and peaks at the seventh decade. By contrast, uterine leiomyosarcoma occurs from the third decade into old age, but is most common in the perimenopausal age group, in the fifth decade. The sex incidence depends on tumor location, with most patients with retroperitoneal and inferior vena cava leiomyosarcoma being women, whereas there is a mild male predominance in noncutaneous soft tissue sites and cutaneous leiomyosarcoma.
Causes and Predisposing Factors
There are few clear causal or predisposing factors identified for this disease. Several exogenous agents have been studied, but only Epstein-Barr virus (EBV) infection, in the setting of severe immunosuppression, has been associated with leiomyosarcomas among patients with acquired immunodeficiency syndrome (AIDS) and after kidney, cardiac, and liver transplantation. Most of EBV-related leiomyosarcomas occur in children and young adults, and develop in organs not traditionally considered preferred sites for leiomyosarcoma. Other traditional risk factors for sarcomas, such as radiotherapy, rarely lead to the development of leiomyosarcomas. In addition, the role for estrogenic stimulation remains unclear. Patients with hereditary retinoblastoma have a cumulative risk of 13.1% for developing any soft tissue sarcoma as a secondary malignancy, including leiomyosarcoma, which further agrees with the relevance of RB1 loss in sporadic leiomyosarcoma (discussed later). In contrast, although clearly reported, leiomyosarcoma is an uncommon subtype of sarcoma among patients with Li-Fraumeni syndrome.
There is no solid evidence showing that leiomyoma (the benign counterpart of leiomyosarcomas) can undergo malignant transformation, and it is currently widely accepted that leiomyosarcomas are tumors that arise de novo.
Pathology and tumor biology
Histopathology
Leiomyosarcoma is a malignant mesenchymal tumor composed of cells showing distinct features of the smooth muscle lineage. The typical histologic pattern of leiomyosarcomas of any origin is that of intersecting, sharply marginated fascicles of spindle cells with abundant eosinophilic cytoplasm and elongated and hyperchromatic nuclei ( Fig. 1 ). Large leiomyosarcomas frequently contain coagulative tumor necrosis regions. Focal pleomorphism is common, and some cases show extensive pleomorphism, resembling any undifferentiated soft tissue sarcoma. Most leiomyosarcomas are reactive for alpha smooth muscle actin, desmin, and h-caldesmon on immunohistochemistry, although none of these markers are specific for smooth muscle differentiation.
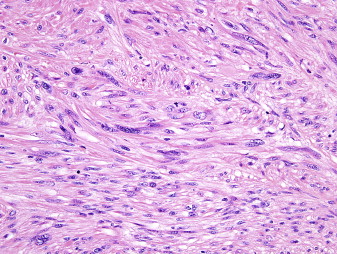
Tumor Biology
From a molecular genetics standpoint, sarcomas are conceptually classified in 2 broad categories : the first category comprises sarcomas with near-diploid karyotypes and simple but essential genetic alterations, such as translocations or activating mutations. The second category includes tumors with complex and unbalanced karyotypes, which are characteristic from tumors with severe genome instability, thus resulting in multiple genomic aberrations, and leiomyosarcomas are in this second category ( Fig. 2 ).
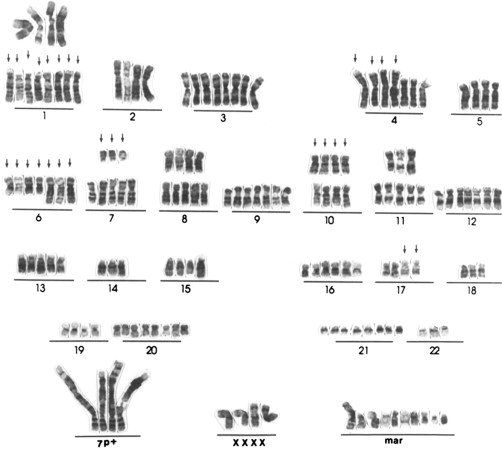
Standard karyotyping and fluorescence in situ hybridization techniques have shown that the cytogenetic and molecular changes in leiomyosarcomas are complex and there are no consistent, recurrent aberrations shown at the chromosomal level. DNA copy number changes also depict a complex landscape in all leiomyosarcomas, and likewise some other cancer types, and the extent of gains/losses and cytogenetic changes seems to be associated with tumor evolution and worse survival. The most consistent changes detected across several studies are losses in chromosomes 10q11 to 21.2 and 13q14.3 to q21.1, and gains at 17p11 to p12. Regions deleted in 10q and 13q harbor 2 important tumor suppressor genes: RB1 and PTEN , respectively.
The underlying genetic mechanisms of sarcomas with complex karyotypic defects, such as leiomyosarcomas, although poorly understood, frequently include disturbances in key cell-cycle genes. The aforementioned 13q loss specifically targets the tumor suppressor RB1 gene, and entails lack of regulation of the cell cycle at the G1-S checkpoint, thus leading to uncontrolled cell division. Analysis of components of the RB1-cyclin D1 pathway (RB1, CDKN2A, CCND1, CCND3) in leiomyosarcomas revealed alterations in up to 90% of patients and association with poorer prognosis (reviewed by Yang and colleagues in 2009). Therefore, loss of RB1 function may be an important driver for proliferation in at least a subset of leiomyosarcoma. In contrast, molecular studies focused on p53/MDM2 abnormalities in leiomyosarcoma have observed a lower rate of p53 mutations and amplification of MDM2 compared with other sarcoma types.
From a gene expression standpoint, gene expression profiling studies using expression microarrays have identified 3 reproducible molecular subtypes that are distributed similarly over leiomyosarcomas of gynecologic and nongynecologic origins. Group I comprises approximately 25% of all leiomyosarcomas, is highly enriched for genes related to muscle contraction and the actin cytoskeleton, tends to be of the conventional leiomyosarcoma histologic subtype, and shows improved outcome compared with leiomyosarcoma groups II and III, which in turn overlaps to a greater extent with undifferentiated pleomorphic sarcomas. These results agree with previous data showing that some leiomyosarcomas cluster with undifferentiated pleomorphic sarcomas and liposarcomas in microarray studies performed in pooled sarcoma subtypes. Profiling studies have also contributed to the identification of new targets, such as Aurora-A and Aurora-B kinases, which are consistently overexpressed in uterine leiomyosarcoma, and in vitro and in vivo targeting of Aurora-A kinase induces cell-cycle arrest and apoptosis.
Several pathways and signal intermediates have been investigated in leiomyosarcoma, and the relevance of phosphatidylinositol 3 kinase (PI3K)/AKT pathway activation has been consistently shown throughout several studies. Genomic deletion of chromosome 10q targets the PTEN tumor suppressor gene and leads to hyperactivation of PI3K/AKT, which is a common finding in leiomyosarcoma. Mice with genetic inactivation of PTEN in the smooth cell muscle lineage accordingly develop a rapid onset of abdominal leiomyosarcomas with constitutive mammalian target of rapamycin (mTOR) activation. Although approximately half of leiomyosarcomas express insulinlike growth factor (IGF)-1R and IGF-II, the relevance of the IGFR/AKT pathway in the proliferation and survival of leiomyosarcoma is yet to be elucidated.
Principles of general management
Diagnosis
Clinical presentation of leiomyosarcomas, as of other soft tissue sarcomas, is often associated with nonspecific symptoms caused by displacement of structures, rather than invasion, in specific anatomic locations of the primary tumor and its metastasis. Pretreatment biopsy is mandatory, but further pathologic evaluation with the primary tumor is typically performed following complete resection.
Imaging approaches include magnetic resonance imaging (MRI) in soft tissue tumors, and contrast-enhanced computed tomography (CT) scan for retroperitoneal lesions. Chest and abdominal CT scan is required in the initial work-up, because hematogenous spread is a frequent event in leiomyosarcomas, and lung and liver are two common sites of metastasis.
Prognostic Factors
In leiomyosarcoma, as in soft tissue sarcomas, histologic grade, tumor size, and tumor depth are the three major clinicopathologic factors that establish the risk profile, and all are included in the American Joint Committee on Cancer (AJCC) staging system for soft tissue sarcomas. However, relationship between AJCC staging and prognosis of leiomyosarcoma arising from the gastrointestinal tract or uterus is not clear. Perhaps the major limitation of the AJCC staging system is that it does not include histology and anatomic site, and both have been shown to affect prognosis. Pisters and colleagues reported the risk factors associated with soft tissue sarcoma in a series of 1,041 cases. Among several risk factors, patients with leiomyosarcoma were at higher risk for distant recurrence and decreased disease-specific survival than other histotypes, which corroborates the intrinsic aggressive behavior of leiomyosarcoma.
Grade and location are two independent prognosis factors of similar importance to stage. Histologic grading is an independent indicator of the degree of malignancy, probability of distant metastasis, and disease-specific survival, and, accordingly, leiomyosarcoma is a subtype of soft tissue sarcoma with substantial intrinsic aggressiveness: approximately 90% of leiomyosarcoma are reported to be moderate to high grade. Regarding location, soft tissue sarcomas from extremities have better overall survival than those arising in the retroperitoneum. Atypical intradermal smooth muscle neoplasm (formerly called cutaneous leiomyosarcoma) constitutes a distinctive entity with excellent prognosis, because it arises in the dermis, and does not develop metastasis. Tumor size and bone or neurovascular involvement are two other risk factors, together with grade, that have been significantly associated with poor outcome specifically in leiomyosarcoma.
Surgery
Surgical resection is the cornerstone treatment of all patients with localized soft tissue sarcomas, and subsequently for leiomyosarcomas. The standard surgical procedure involves a complete excision with wide negative margins (R0 resection), which offers the best chance of cure, with or without adjuvant treatment.
Leiomyosarcomas are commonly found in the retroperitoneum, and surgical R0 resection in retroperitoneal leiomyosarcoma is often hampered by the large tumor size, coupled with the anatomic constrains. Thus, in clinical practice, many resections are grossly complete but with microscopically positive margins, which is essential because the ability to perform a complete surgical resection at the time of initial presentation is the most important prognostic factor for survival.
In addition, surgery may benefit certain metastatic patients, particularly pulmonary metastasectomy in patients with a low number of metastases appearing late after primary resection.
Radiotherapy
The therapeutic role of radiotherapy in soft tissue sarcomas has been shown to improve local control with preservation of the function, decrease in local recurrence rate, but not to improve overall survival. Thus, preoperative or postoperative radiotherapy are considered to be the standard of care for nearly all intermediate-grade or high-grade leiomyosarcomas of the limbs and trunk. The radiotherapy approach in soft tissue sarcomas is discussed in detail elsewhere.
Radiation therapy has shown benefit in the treatment of soft tissue sarcoma of the extremity and trunk, but there are no randomized data addressing this question in retroperitoneal sarcomas and hence there is institutional variation on its use in this setting. The largest retrospective series investigated the role of preoperative radiation therapy in 33 retroperitoneal sarcomas, 12 of which were leiomyosarcomas. In this series, leiomyosarcomas, unlike liposarcomas, had a lower 3-year local recurrence rate (18.8%) and a higher 3-year distance recurrence rate (35.4%).
Systemic treatment of leiomyosarcomas
The most life-threatening aspect of sarcomas in general, and leiomyosarcomas in particular, is their propensity for hematogenous dissemination, and therefore systemic control is desired.
The role of postoperative chemotherapy remains unproven, and is not further discussed here because it is not the focus of this article. Most trials evaluating adjuvant chemotherapy in sarcoma evaluate patients with high-risk soft tissue sarcomas and including many histologic subtypes, making it difficult to discern a subtype-specific recommendation. The largest such trial was recently reported by the European Organisation for Research and Treatment of Cancer (EORTC) and did not show an overall survival benefit with adjuvant chemotherapy.
Almost all patients with unresectable metastatic leiomyosarcoma are considered incurable; hence, in almost all cases, the treatment intention for systemic disease is palliative, with the goal of decreasing tumor bulk, diminishing symptoms, improving quality of life, and prolonging survival. It is widely recognized that different histologic subtypes have variable patterns of chemosensitivity, and leiomyosarcomas show moderate sensitivity to chemotherapy (reviewed by Grimer and colleagues in 2010), although uterine leiomyosarcoma seems to be more responsive to many chemotherapy agents, in contrast with visceral leiomyosarcomas. Systemic treatment of uterine leiomyosarcoma is described separately below.
Anthracyclines
Single-agent doxorubicin is a standard systemic treatment of soft tissue sarcomas, and subsequently for leiomyosarcomas as well. Doxorubicin, as a single agent, has reported response rates varying between 10% and 25% (reviewed by Krikelis and Judson in 2010), although leiomyosarcoma seems to be less responsive than other sarcoma subtypes, such as synovial sarcoma and liposarcoma. A review of 2185 patients with sarcoma treated with doxorubicin showed that leiomyosarcomas had a nonsignificant lower response rate (11%) compared with other sarcoma subtypes. An updated study on 488 metastatic patients, 85% of whom received doxorubicin as first-line treatment and including 171 leiomyosarcomas, also concluded that synovial sarcoma and liposarcoma were independent favorable predictive factors of response and longer survival compared with leiomyosarcomas.
Various doxorubicin-based combination regimens have been tested in the hopes of achieving superior response rates and overall survival, and higher response rates (of approximately 45%) have been consistently shown in several randomized clinical trials and pooled analyses with combination regimens compared with single-agent doxorubicin. However, none of these studies have been able to show whether the observed increased activity leads to a statistically significant advantage in overall survival. In addition, more toxic effects were observed with the combination regimens. Therefore, the higher response rates suggest that doxorubicin/ifosfamide might be justified in selected patients and if tumor shrinkage is critical.
In addition, alternative anthracyclines might be used to minimize secondary effects, such as epirubicin and liposomal anthracyclines. Epirubicin is less cardiotoxic and provides outcomes that are comparable with doxorubicin. Liposomal doxorubicin has a better toxicity profile, although whether it is as efficacious as unencapsulated doxorubicin is still unproved in large trials.
Ifosfamide
Single-agent ifosfamide seems to have similar antitumor activity to doxorubicin in soft tissue sarcomas, but entails a worse toxicity profile, and thus may be regarded as a second-line regimen. Treatment with ifosfamide among patients who previously failed a doxorubicin-based regimen achieves approximate response rates of 25%, with an observed dose-response relationship.
Sleijfer and colleagues undertook a retrospective analysis on prognostic and predictive factors for outcome to first-line single-agent ifosfamide and ifosfamide-containing regimens in 1337 advanced or metastatic patients with soft tissue sarcoma. The median progression-free and overall survivals were 19 and 54 weeks respectively. There was a nonsignificant trend in leiomyosarcomas toward a lower response rate and a lower median progression-free survival compared with synovial sarcomas, liposarcomas, and other sarcoma histologies (not including gastrointestinal stromal tumors).
Gemcitabine and Gemcitabine-based Regimens
Gemcitabine is a nucleoside metabolite that has shown efficacy in a variety of solid tumors, and several phase II clinical trials have shown a response rate of approximately 10% in previously treated soft tissue sarcomas, including leiomyosarcomas.
Several other agents have been tested in soft tissue sarcomas, and none, as single therapy, has achieved response rates greater than 20%. The activity of docetaxel and vinorelbine as single agents is almost nill. Dacarbazine and temozolomide seem to have modest antitumor effects, with some predilection for leiomyosarcomas, and data from a phase II clinical trial showed an overall response rate of 15.5%. The combination of gemcitabine with any of these aforementioned drugs seems to yield significant synergistic activity, and a gemcitabine/docetaxel doublet has been shown to be highly active in patients with unresectable leiomyosarcomas or in those with leiomyosarcoma that has progressed to prior doxorubicin-based therapy.
Activity for fixed-dose rate gemcitabine plus docetaxel was initially reported in a prospective phase II study that recruited patients with advanced leiomyosarcoma of uterine (n = 29) or other (n = 5) primary sites progressing to prior treatment. Complete response was observed in 3 patients and partial response in 15, for an overall response rate of 53%. Among 16 patients previously treated with doxorubicin with or without ifosfamide, objective responses were observed in 8 patients (50%). Among the 5 patients with nonuterine primary leiomyosarcoma, partial responses were observed in 2 patients (40%), stable disease in 2, and progression of disease in 1. The median progression-free survival was 5.6 months. The activity of the combination and the activity in leiomyosarcoma were later confirmed in 2 independent series of soft tissue sarcomas.
It was unknown whether adding docetaxel produces synergy in the combination, or whether most of the treatment effect arises from the fixed dose rate of gemcitabine. Two multicenter phase II randomized clinical trials addressed this question. The SARC002 study showed that the combination was associated with superior response rate (16% vs 8%), median progression-free survival (6.2 vs 3.0 months), and overall survival (17.9 vs 11.5 months) compared with gemcitabine alone in patients with advanced soft tissue sarcoma who had received a median of 1 line of treatment. This study also confirmed the higher sensitivity of leiomyosarcoma to this regimen. By contrast, results from the French clinical trial differed from previous data and only confirmed the benefit of the combination in uterine leiomyosarcomas, but not in those of nonuterine origin, in which the combination seemed to be detrimental. The French study, unlike the SARC02 trial, included only patients with leiomyosarcomas after progressing to a first line of systemic treatment, and the dose of gemcitabine in the control arm was slightly higher. Whether these nuances might explain the differences observed between the current conflicting data remains unknown.
Two other gemcitabine-based combinations have shown benefit in soft tissue sarcomas, and exquisite efficacy in leiomyosarcomas. García del Muro and colleagues explored the feasibility and activity of fixed-dose-rate gemcitabine plus dacarbazine in a phase II randomized clinical trial. The combination was superior to single-agent dacarbazine, and histology showed that leiomyosarcomas of any origin benefited significantly from the doublet, achieving a median progression-free survival and an overall survival of 4.9 and 18.3 months respectively (2.1 and 7.8 months respectively in nonleiomyosarcoma subtypes). The combination of gemcitabine and vinorelbine similarly achieved greater benefit in leiomyosarcomas, although the complete benefit rate was lower, and the trial did not have a control arm. Table 1 summarizes gemcitabine-based phase II clinical trials.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


