- 9.1 Introduction
- 9.2 Clinical syndromes
- 9.3 Classifications of glomerulonephritis
- 9.3.1 Histological classification
- 9.3.2 Mechanisms of glomerulonephritis
- 9.3.1 Histological classification
- 9.4 Asymptomatic haematuria
- 9.4.1 IgA nephropathy
- 9.4.2 Henoch–Schönlein nephritis
- 9.4.1 IgA nephropathy
- 9.5 Acute glomerulonephritis
- 9.5.1 Acute immune-complex nephritis
- 9.5.2 Acute post-infectious glomerulonephritis
- 9.5.1 Acute immune-complex nephritis
- 9.6 Chronic glomerulonephritis
- 9.6.1 Chronic immune-complex glomerulonephritis
- 9.6.2 Membranoproliferative glomerulonephritis (mesangiocapillary glomerulonephritis)
- 9.6.3 Lupus nephritis
- 9.6.1 Chronic immune-complex glomerulonephritis
- 9.7 Rapidly progressive glomerulonephritis
- 9.7.1 Anti-glomerular basement membrane disease
- 9.7.2 Antineutrophil cytoplasmic antibody-associated glomerulonephritis
- 9.7.1 Anti-glomerular basement membrane disease
- 9.8 Nephrotic syndrome
- 9.8.1 Minimal-change nephropathy
- 9.8.2 Focal glomerulosclerosis
- 9.8.3 Membranous glomerulonephritis
- 9.8.4 Amyloid disease
- 9.8.5 Other causes of nephrotic syndrome
- 9.8.1 Minimal-change nephropathy
- 9.9 Tubulointerstitial nephropathy
- 9.9.1 Acute drug-induced tubulointerstitial nephritis
- 9.9.2 Multiple myeloma and myeloma kidney
- 9.9.3 Other immunologically mediated tubulointerstitial nephritides
- 9.9.1 Acute drug-induced tubulointerstitial nephritis
- 9.10 Chronic renal failure
- 9.11 Recurrent glomerulonephritis in transplanted kidneys
 Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
9.1 Introduction
Renal disease includes damage to the glomeruli, the tubules or the interstitial tissue. Immunological components are involved in most cases of glomerular damage (glomerulonephritis) and for some forms of injury to the renal tubules and interstitium (tubulointerstitial nephritis), although the precise mechanisms are not always clear. Humoral and cell-mediated immune mechanisms (Box 9.1a,b) play a part in the pathogenesis of nephritis but some common types of glomerulonephritis (e.g. minimal-change disease) and tubulointerstitial disease do not have a clear-cut immune basis.
- Antibody may react directly with the glomerular or tubular basement membrane
- Antibody may form immune complexes with antigens that subsequently lodge in the kidney
- Antigen may bind with, or be trapped in, the glomerular basement membrane and react with antibody subsequently
- Antibody may induce a vasculitic process that damages the capillary plexus of the glomerulus
- Adoptive transfer of sensitized T cells to rats treated with sub-nephritogenic doses of antibody causes glomerular hypercellularity due to proliferation of resident glomerular cells and an influx of mononuclear leucocytes.
- Severe proliferative nephritis can develop after immunization with glomerular basement membrane in bursectomized chickens unable to mount antibody responses
- In humans, T lymphocytes have been found in proliferative and non-proliferative glomerulopathies
- Treatment with ciclosporin is effective in some glomerular disorders
The terminology of glomerulonephritis is confusing because three descriptive classifications have been in simultaneous use for many years, but none is entirely satisfactory in isolation. There is a clinical classification, describing the commoner modes of presentation; a morphological classification, based on light and electron microscope findings; and an immunological classification, based on the proposed immune mechanism of renal damage.
9.2 Clinical syndromes
Several clinical syndromes are recognized, but with considerable overlap.
Recurrent haematuria may be the first manifestation of renal or extrarenal disease. It can be macroscopic or microscopic. Haematuria of unknown origin requires urological investigation to exclude a site of bleeding in the upper and lower urinary tracts, particularly since tumours of the bladder are the second most common cause after infections.
Persistent proteinuria: a small amount of albumin (up to 30 mg/day) is normally present in urine of healthy adults. Amounts in excess of this are pathological. Excretion of >300 mg albumin per 24 h is termed ‘overt albuminuria’, while excretion of amounts between 30 and 300 mg/day is called ‘microalbuminuria’. Overt proteinuria is a cardinal sign of established glomerular damage and a risk factor for declining renal function. The higher the level of proteinuria, the faster the development of tubulointerstitial lesions and renal fibrosis progressing to chronic renal failure. Proteinuria is often discovered by chance when urine is tested for some other reason. Microalbuminuria is used as a marker of early diabetic nephropathy and therapeutic interventions are increasingly targeted at the early microalbuminuric stage in the belief that pathological damage may be modifiable or even reversible at this point.
Nephrotic syndrome is defined as low plasma levels of albumin (hypoalbuminaemia) accompanied by dependent oedema resulting from severe proteinuria, usually in excess of 3.5 g/day. In children, this is a common presentation of glomerulonephritis.
Acute nephritis is characterized by sudden onset of haematuria, proteinuria, hypertension and oliguria.
Renal failure may be acute or chronic. Acute renal failure is a period of sudden, severe impairment of renal function, usually triggered by some vascular or inflammatory insult. Chronic renal failure may be the end result of any disorder that destroys normal renal architecture; although there are many causes, about half of cases are caused by some type of glomerulonephritis.
There is a poor correlation between the clinical picture and the underlying morphology. A specific form of glomerulonephritis can show different clinical features in different patients or even in the same patient at different times (Fig. 9.1). A definite diagnosis, allowing for more accurate assessment of prognosis, is best made by renal biopsy, the results of which tie up with patient outcome in terms of disease progression.
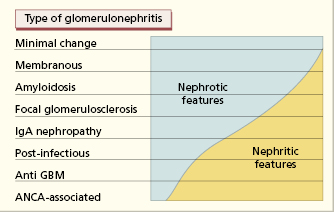
9.3 Classifications of glomerulonephritis
An understanding of glomerulonephritis requires a grasp of normal glomerular structure (Fig. 9.2a,b). The glomerulus is a unique capillary plexus, fed by an afferent arteriole and drained by an efferent arteriole, supported by a stalk called the mesangium. As the afferent arteriole enters the glomerulus, it divides into numerous capillary loops. The wall of the capillary loop (Fig. 9.3) acts as the glomerular filter and is composed of three layers: an inner layer of glomerular endothelial cells, an outer (i.e. urinary side) layer of glomerular epithelial cells, and the glomerular basement membrane between them. Each layer has specialized features distinguishing it from capillary walls elsewhere in the body.
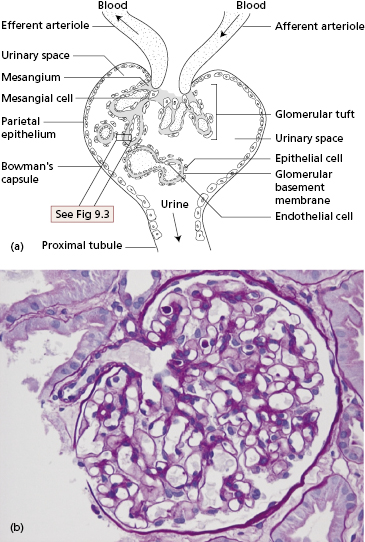
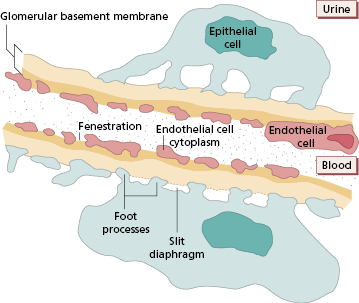
Endothelial cells offer no anatomical barrier to the passage of molecules. The cytoplasm of the endothelial cells forms a thin layer perforated by fenestrations much larger in diameter than macromolecules in the plasma. These openings allow capillary blood to come into direct contact with the glomerular basement membrane.
Glomerular basement membrane (GBM) has unique constituents with restricted isoforms of laminin, type IV collagen and proteoglycans, highly negatively charged molecules that account for the charge-dependent filtration that normally conserves plasma proteins.
Epithelial cells or podocytes are arranged around the capillaries (Fig. 9.3). Epithelial cells are phagocytic. Each cell has multiple foot processes which interdigitate and are partly embedded in the outer layer of the GBM. A thin membrane – called the slit diaphragm – bridges the spaces between the foot processes of the podocytes and is vital for maintaining the barrier to plasma proteins. Several interacting slit diaphragm proteins have been identified and mutations in the genes coding for some of them are associated with inherited forms of nephrotic syndrome, strongly implying that podocytes are vital in preventing proteinuria in healthy people. Disruption of structural slit diaphragm proteins by proteinuria may predispose to apoptosis (death) of podocytes.
The mesangium, the central core of tissue in the glomerulus, is made up of mesangial cells separated by an extensive matrix. The mesangial region can take up and dispose of large molecules, particularly immune complexes. The mesangial matrix ultimately drains into renal lymphatics and so, via para-aortic lymph nodes, to the venous blood supply.
9.3.1 Histological classification
Glomeruli are composed of three main cell types – mesangial, endothelial and epithelial – and any or all of these cells may increase in number in response to injury. The term glomerulonephritis must therefore be qualified (Table 9.1) by defining the types of cells affected and whether part or all of the glomerulus is damaged. Classification is based on light microscopy, electron microscopy and immunohistochemistry.
Table 9.1 Some descriptive terms used in the morphological classification of nephritis
| Extent of damage | |
| Involving all glomeruli |
| Involving some glomeruli only |
| Involving part of a glomerulus while the rest of that glomerulus appears normal |
| Cellular changes | |
| An increase in the numbers of cells within the glomerular tuft. Subgroups exist in which proliferation is predominantly confined to a particular cell type |
| Thickening of the glomerular capillary wall by abnormal deposits on the epithelial aspect of the basement membrane |
| Proliferation of cells plus thickening of the glomerular capillary wall |
| Proliferation of parietal epithelial cells (extracapillary proliferation) |
9.3.2 Mechanisms of glomerulonephritis
The evidence that human glomerulonephritides are immunological disorders relies heavily on animal experiments. At least two mechanisms are known to induce glomerulonephritis in animals:
- deposition of circulating antigen–antibody complexes within glomeruli – ‘immune-complex nephritis’, an example of type III hypersensitivity (see Chapter 1); and
- reaction of circulating antibodies with antigens which are either part of the GBM (for instance, ‘anti-GBM’ nephritis), antigens that have been trapped there or antigenic components of renal vascular endothelium (Fig. 9.4), examples of type II hypersensitivity.
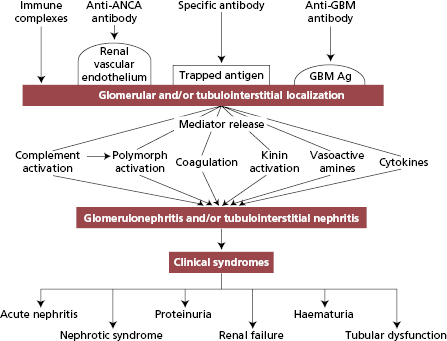
After the initiation of glomerular injury, a number of proinflammatory mediator pathways (Fig. 9.4) are activated both in infiltrating cells and in resident glomerular cells and participate in the destructive and restorative processes. Remodelling of extracellular matrix after injury generates signals that differ from those transmitted by normal glomerular matrix and induces activation and proliferation of resident and infiltrating cells in the glomerulus. Haemodynamic changes cause hyperfiltration, intraglomerular hypertension and abnormal intravascular shear forces that can worsen glomerular injury. Depending on the cells affected, apoptosis may have a crucial role either in resolution of damage or in causing glomerular scarring.
9.4 Asymptomatic haematuria
Some forms of glomerulonephritis often follow infections of the respiratory tract and usually involve mesangial deposits of IgA.
9.4.1 IgA nephropathy
IgA nephropathy (‘mesangial IgA deposition’ or ‘Berger’s disease’) is the most common form of primary glomerulonephritis in the world. It accounts for about 10% of all cases of primary glomerular disease in the USA, 20% of cases in Europe and 30–40% in Asia. It affects mainly older children or young adults, is often asymptomatic (a chance finding of microscopic haematuria) but may present as recurrent episodes of macroscopic haematuria occurring after an upper respiratory tract infection (Case 9.1) or, less frequently, a gastrointestinal or urinary tract infection, or strenuous exercise; presentation with acute nephritis, hypertension or nephrotic syndrome is less frequent. In contrast to post-streptococcal glomerulonephritis (see Case 9.3), the period between infection and haematuria is short, ranging from hours to a few days.
 Case 9.1 IgA nephropathy
Case 9.1 IgA nephropathyA 14-year-old boy presented with an 18-month history of intermittent, painless haematuria, usually occurring after strenuous exercise, but without dysuria or increased frequency of micturition. He also had frequent colds and sore throats and believed that the haematuria also happened at these times. On examination, he appeared fit and healthy; his blood pressure was 120/75. Urine analysis showed microscopic haematuria (3+) and a trace of protein. Intravenous urography, a micturating cystogram and cystoscopy were normal. His haemoglobin, white cell count, blood urea and creatinine clearance were normal; the urinary protein excretion was 0.95 g/day. Immunoglobulin, CH50, C4 and C3 levels were within normal limits. In view of the duration of haematuria, a renal biopsy was performed. Twelve glomeruli were present: all showed a diffuse increase in mesangial cells with thickening of the matrix. Immunofluorescent examination of the biopsy showed mesangial deposits of IgA and C3 (Fig. 9.5a). The appearances were characteristic of IgA nephropathy.
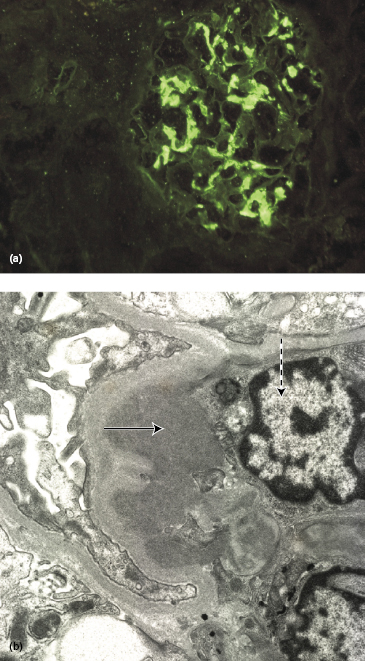
The clinical features are variable and yet the biopsy findings are constant and probably persist indefinitely. On light microscopy, the glomeruli show focal and segmental mesangial proliferation and, as the name of the condition implies, prominent deposits of IgA are found in the mesangium of every glomerulus (Fig. 9.5a,b), together with complement components of the alternate pathway.
Fig. 9.5 (a) IgA nephropathy showing IgA deposits in the mesangium on immunofluorescence. (b) Electron micrograph of IgA nephropathy showing an IgA deposit (arrowed) and an adjacent mesangial cell (interrupted arrow).
In terms of pathogenesis, IgA nephropathy can be considered a type of renal limited vasculitis caused by an innate defect in IgA mucosal immunity in the gut or lung: repeated exposure to a variety of environmental antigens results in an abnormal IgA response, namely the generation of nephritogenic polymeric IgA antibodies with defective galactosylation of the IgA hinge region resulting in deposition in the mesangium and the induction of inflammation in genetically susceptible individuals. Genome-wide association studies (GWAS) have shown a number of genetic polymorphisms relating to susceptibility such as those involved in the response to pathogens [toll-like receptor (TLR)-9 polymorphisms], the response to IgA immune deposits (FcγR 2a, 3a and 3b polymorphisms) or the activity of the renin–angiotensin pathway [angiotensin-converting enzyme (ACE) polymorphisms]. Clinical evidence includes an association between IgA nephropathy and chronic liver disease, coeliac disease and dermatitis herpetiformis, other disorders that are also linked with immune complexes containing IgA.
Predictors for prognosis are being sought actively but currently depend on the biopsy and clinical findings. Patients presenting with nephrotic-range proteinuria, hypertension or crescents on biopsy are more likely to progress to renal failure. Spontaneous clinical remission occurs in about 10% of patients. Of the remainder, renal survival of 80–90% at 10 years and 50–70% at 20 years implies that IgA nephropathy can be a relatively benign disease with a good prognosis but, because IgA nephropathy is so common, it contributes significantly to the population with end-stage renal failure (ESRF) (30% of cases). The risk of developing renal failure increases by about 1% per year.
Renal transplantation has provided the strongest support for IgA nephropathy as a systemic disease. Recurrence in the donor kidney can be demonstrated in around one-third of patients who receive a transplant for ESRF due to IgA nephropathy. Further, when a kidney is inadvertently sourced from a patient with asymptomatic IgA nephropathy and transplanted to a recipient, histological evidence of IgA nephropathy quickly disappears.
There is no specific treatment and trials of immunosuppression and plasma exchange have been controversial. Treatment is limited to those with a poor long-term prognosis. Angiotensin II receptor blockers, ACE inhibitors and fish oil supplements have been used for their protective and antiproteinuria effects, with some benefit.
9.4.2 Henoch–Schönlein nephritis
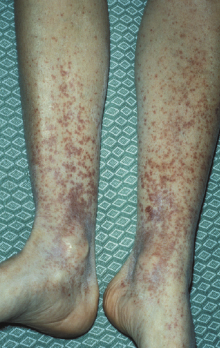
Henoch–Schönlein nephritis (Henoch–Schönlein purpura or anaphylactoid purpura) is a common form of systemic vasculitis in which small blood vessels in a number of organs are involved. It is usually a disease of children, with a peak age of onset between 4 and 10 years. The syndrome is characterized by non-thrombocytopenic purpura of the skin (particularly around joints) (Fig. 9.6), arthralgia, gastrointestinal pain and glomerulonephritis as in Case 9.2. Kidney disease is the most important manifestation of HSP as renal failure is the main cause of death. The overall prevalence of renal disease varies from 40% to 100% but in most patients this is mild; progression to renal failure occurs in fewer than 10%. Those with the most severe clinical presentation have the worst outcome: about 40% of those with nephritic or nephrotic syndromes at onset show long-term impairment of renal function. Treatment is largely empirical, controversial and, at best, only partially effective. Steroids seem to control the joint and abdominal pain but have no effect on the skin or renal involvement at routinely used doses. Despite the uncertainty regarding therapy, the short-term prognosis is favourable. However, recurrence of HSP is common.
 Case 9.2 Henoch–Schönlein nephritis
Case 9.2 Henoch–Schönlein nephritisA 12-year-old boy presented with a 1-week history of pain in the left loin. This was diagnosed as a urinary tract infection and treated with amoxicillin. One week later, he developed a purpuric rash around the ankles, accompanied by some blistering and superficial necrosis. Shortly afterwards, he developed pain in the left elbow joint. On admission to hospital, he was noted to have haematuria and proteinuria and a blood pressure of 130/90. Over the next month, he suffered further episodes of abdominal colic and purpura. His haemoglobin was 95 g/l with a normal white cell count. Antinuclear antibodies were negative and total haemolytic complement, C4 and C3 levels were normal. Although his blood urea was normal, his creatinine clearance was low at 31 ml/min per m2 with proteinuria of 4.5 g/day.
A skin biopsy of a purpuric lesion showed vasculitic changes in the dermis, with IgA and C3 deposition in the blood-vessel walls. A renal biopsy, containing 21 glomeruli per section, showed epithelial crescents and diffuse mesangial hypercellularity in seven glomeruli. On immunofluorescence, granular deposits of IgA and C3 and, to a lesser extent, IgG and properdin were present in the mesangium. The clinical and histological features were those of Henoch–Schönlein nephritis (HSN). Because of the heavy proteinuria and diminished creatinine clearance, he was treated with a limited course of corticosteroids. Over a 9-month follow-up period, the purpura and episodes of abdominal colic subsided, his creatinine clearance increased to 47 ml/min, but he continued to have moderately heavy proteinuria (3.2 g/day). The prognosis is uncertain.
Immunohistology of the renal biopsy shows irregular, granular deposits of IgA, C3 and fibrin in the glomeruli. Deposits of IgA and C3 are also found in the skin, even in non-affected areas, and are diagnostic of the condition. As in IgA nephropathy, the available evidence suggests an IgA-dominant immune-complex pathogenesis with complement activation occurring via the alternate pathway. A variety of bacterial or viral antigens could be involved, as there is an association with preceding upper respiratory tract infection. In addition, HSN is a seasonal disease: most patients present during the winter. The clinical and immunological similarity between HSN and IgA nephropathy suggests that IgA nephropathy is a renal limited form of HSN.
9.5 Acute glomerulonephritis
9.5.1 Acute immune-complex nephritis
A single intravenous injection of a foreign protein into a rabbit causes vasculitis, arthritis and glomerulonephritis about 10 days later (‘one-shot’ serum sickness) (Fig. 9.7). This occurs when the amount of circulating antigen is still in excess of specific IgG antibody produced in response to the stimulus (see Chapter 1); the small immune complexes so formed are soluble but become trapped in capillary membranes, particularly in the kidney. Immunofluorescent examination of the kidney shows deposition of the injected antigen, specific antibodies and complement components in an irregular, granular (‘lumpy-bumpy’) distribution along the GBM. Renal injury is due to the resultant attraction and accumulation of polymorphs in the glomeruli and release of inflammatory mediators.
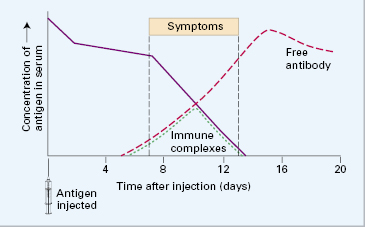
The symptomatic phase is usually transient and subsides, with complete healing, as the complexes, both soluble and fixed, are cleared in around 2–4 weeks.
9.5.2 Acute post-infectious glomerulonephritis
In adults, post-infectious glomerulonephritis is seen increasingly in immunocompromised adults and in elderly people. It is frequently linked to staphylococcal and Gram-negative bacterial infections, often at multiple sites. The clinical presentation can be insidious and the diagnosis made only after renal biopsy suggests an infectious cause. In contrast to post-streptococcal disease, the destructive glomerular proliferation often persists and the prognosis is poor. Post-infectious glomerulonephritis may also follow parasitic (malaria, filariasis) and viral (hepatitis B or C) infections (Case 9.3).
 Case 9.3 Post-streptococcal glomerulonephritis
Case 9.3 Post-streptococcal glomerulonephritisA 9-year-old boy was admitted as an emergency with puffiness of the face, eyes and trunk. A week previously he had complained of a sore throat. On examination, he was mildly pyrexial (temperature 37.5°C) and hypertensive (BP 170/110). There was periorbital and scrotal oedema. His urine showed proteinuria, haematuria and red cell casts. He was anaemic (Hb 107 g/l) with a normal white cell count and differential. A throat swab grew normal flora but antibodies to streptococcal antigens were present in high titre: antistreptolysin O titre 1600 IU/ml (normal <300 IU/ml). Serum complement studies done 3 days after admission showed a very low C3 (0.10 g/l; NR 0.8–1.40) and a normal C4 (0.23 g/l; NR 0.2–0.4). His creatinine clearance was 46 ml/min, serum albumin 29 g/l and urinary protein excretion 1.5 g/day.
These findings were typical of post-streptococcal glomerulonephritis and so renal biopsy was not performed. As anticipated, the serum complement returned to normal in 4 weeks, accompanied by disappearance of the proteinuria and hypertension, although a small amount of microscopic haematuria persisted. The prognosis is good. An unusual feature of this case was the degree of hypertension.
Acute post-streptococcal glomerulonephritis (PSGN) is mainly seen in countries in which antibiotics for streptococcal infections are not widely available and accounts for about a third of cases of acute GMN. It is a disease of children aged 2–10 years, but adolescents and adults may be affected. Over 90% of cases are preceded by streptococcal infection of the throat or skin. Patients typically present with acute nephritis 7–12 days after a throat infection or about 3 weeks after a skin infection. The diagnosis of PSGN rests on prior microbiological culture, increasing titres of streptococcal antibodies and a low serum C3 level. Laboratories can often test for a range of antistreptococcal antibodies including antistreptolysin (ASO), antihyaluronidase (AHase), antistreptokinase (ASKase), antinicotinamide-adenine dinucleotidase (anti-NAD) and anti-DNAse B antibodies. These antibodies are useful in approx. 95% of cases following pharyngitis and 80% in those following pyoderma.
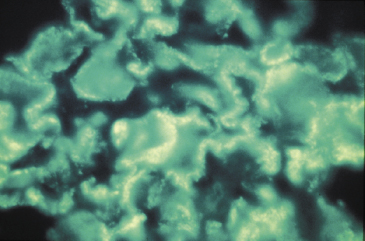
The histological features depend on the timing of biopsy. Acute post-streptococcal glomerulonephritis is characterized by the presence of electron-dense deposits (‘humps’) on the epithelial side of the GBM (Fig. 9.8a,b): these represent the discrete ‘lumpy-bumpy’ deposits of IgG and C3 found by immunofluorescence along the capillary loop in sites corresponding to the ‘humps’ (Fig. 9.9). There is also diffuse proliferation of endothelial and mesangial cells and polymorph infiltration of the glomerulus. Antigenic fragments from nephritogenic strains of streptococci bind to the GBM, so localizing specific antibody to this site.
Fig. 9.8 (a) Characteristic ‘humps’ seen in poststreptococcal glomerulonephritis. These are localized, epimembranous, electron-dense deposits found in several forms of acute post-infectious glomerulonephritis. (b) Electron micrograph of acute proliferative poststreptococcal glomerulonephritis showing subepithelial ‘humps’ (arrow) (see also Fig. 9.13).
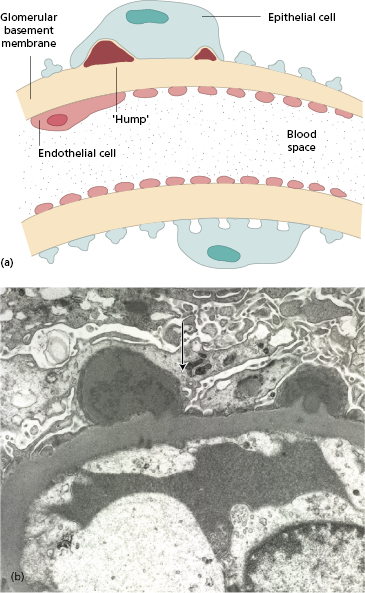
Fig. 9.9 Poststreptococcal glomerulonephritis showing ‘lumpy-bumpy’ deposits on immunofluorescence. Compare this with anti-glomerular basement membrane nephritis (Fig. 9.15b).
The clinical and immunological features of this condition are similar to acute (‘one-shot’) immune-complex nephritis in rabbits. Serum complement C3 is markedly reduced during the early phase, with a gradual return to normal over 6–10 weeks in uncomplicated cases. A low C3 persisting beyond 12 weeks suggests a different diagnosis (see section 9.6.2 and Case 9.4).
The prognosis of acute post-streptococcal glomerulonephritis is good in children, worse in adults. Almost all preschool children will recover, with less than 1% developing crescentic glomerulonephritis.
9.6 Chronic glomerulonephritis
9.6.1 Chronic immune-complex glomerulonephritis
Immune-complex nephritis is believed to account for the majority of cases of human glomerulonephritis, but certain criteria should be fulfilled for complexes to be considered relevant to the pathogenesis of renal disease (Box 9.2). In practice, however, the diagnosis of immune-complex nephritis usually rests solely on immunofluorescent findings similar to those of experimental models of immune-complex disease.
- Immune complexes are present at the site of tissue damage
- The antigen component of the immune complex is identifiable
- Removal of immune complexes produces clinical improvement
The pathogenesis of experimental immune-complex nephritis is well defined. When rabbits are given repeated intravenous injections of a foreign protein, some develop a chronic progressive glomerulonephritis. Damage depends on producing a state of antigen excess after every injection, which saturates free antibody and generates loads of immune complexes. If animals fail to produce any antibody or, instead, mount a strong humoral response that rapidly eliminates the antigen, they do not develop glomerulonephritis. Affected animals produce non-precipitating, low-affinity antibody that is poor at antigen elimination. Even good antibody producers develop nephritis if the repeated antigen dose is increased to maintain antigen excess.
Reasons for chronic immune-complex disease in humans are not fully understood, but comparisons with this experimental model suggest some specific situations in which this is likely to occur (Box 9.3 and Fig. 9.10). Examples of persistent antigen exposure that give rise to immune-complex nephritis are shown in Table 9.2. Chronic infection is the best-recognized source of prolonged antigen exposure.
- Antigen exposure persists (Table 9.2)
- The host makes an abnormal response
- Local factors, such as C3 receptors or changes in permeability that promote deposition of circulating complexes
- Complexes are made less soluble by defects in complement factors
Fig. 9.10 Factors influencing the development of immune-complex disease. CR1, complement receptor 1.
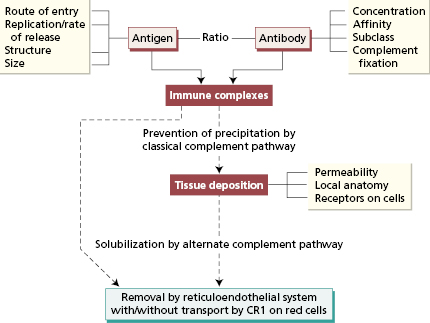
Table 9.2 Examples of immune-complex nephritis in humans
| Antigen* | Associated disease† | |
|---|---|---|
| Exogenous antigens | ||
| Virus | Hepatitis B virus | Hepatitis B |
| Polyarteritis nodosa | ||
| Hepatitis C virus | Mixed essential cryoglobulinaemia | |
| Cytomegalovirus | Glomerulonephritis | |
| Bacteria | Streptococcus | Post-streptococcal glomerulonephritis |
| Streptococcus viridans | Bacterial endocarditis | |
| Staphylococcus | Shunt nephritis | |
| Mycobacterium leprae | Lepromatous leprosy | |
| Parasites | Plasmodium malariae | |