PRACTICE POINTS
- Diabetic ketoacidosis (DKA) is a very serious complication of pregnancy that threatens the mother’s life and the health and viability of the fetus.
- DKA only occurs in situations of severe insulin deficiency combined with increases in catabolic hormones. The metabolic physiology of pregnancy predisposes to DKA.
- Patients present with weakness, hypotension, vomiting, and abdominal pain. The diagnosis is confirmed if there is hyperglycemia, ketonemia, and anion gap acidosis.
- To prevent hypovolemic shock, aspiration, cardiac dysrhythmias, thromboembolism or cerebral edema, prompt management, including fluid and electrolyte replacement, insulin, airway protection, heparin and rigorous monitoring are essential.
- DKA in pregnancy is preventable if monitoring is intensive and corrective action prompt.
CASE HISTORY
A 19-year-old woman with an 11-year history of Type 1 diabetes presented as an emergency with 12 hours of vomiting progressing to drowsiness and collapse. She was at 28 weeks of gestation in her first pregnancy. The pregnancy had been confirmed at 8 weeks of gestation at which time the hemoglobin A1c (HbA1c) was 10.8%. The pregnancy was not planned and it was 2 ½ years since she had last attended anywhere for diabetes review. Her insulin had immediately been changed from twice daily fixed mix to basal bolus and with the help of an intensive re-education program and several times weekly support from the diabetes specialist nurse and midwife, glucose control improved. By the time of the 20-week anomaly scan, which was normal, self-monitoring results were mostly close to target and HbA1c was 6.6%. However, after that visit she failed to attend for her scheduled joint diabetes antenatal clinic review appointments and could not be contacted by the diabetes specialist midwife or her family doctor.
On initial assessment her pulse was 118 bpm, blood pressure 76/42 mmHg, respiratory rate 26 breaths/min, and Glasgow coma score (GCS) 8. There were no pointers to infection. Emergency bloods revealed blood glucose 33.2 mmol/L (603.6 mg/dL), urea 24.2 mmol/L (67.8 mg/dL), creatinine 186 μ mol/L (2.10 mg/dL), sodium 146 mmol/L (146 mEq/L), potassium 5.1 mmol/L (5.1 mEq/L), hemoglobin 13.4 g/L (1.34 g/dL), white cell count (WCC) ‘19.7 x 106/mL, pH 6.94, venous bicarbonate 4 mmol/L (4 mM), base excess -24, pO2 25 kPa (on high-flow oxygen), and pCO2 2.3 kPa. The friend who accompanied her to A & E reported that she had been depressed, had stopped testing her blood glucose, and had been taking her insulin only erratically.
A nasogastric tube was passed and 1.5 L of gastric content drained. One liter of plasma expander was infused over 30 minutes. Blood pressure rose to 90/48 mmHg and pulse reduced to 110 bpm. Urinary catheterization released 200 mL of concentrated urine containing ketones +++.
She was commenced on a standardized DKA protocol involving fluid, insulin, and electrolyte replacement with frequent biochemical monitoring. Tinzaparin 4500 U was started subcutaneously (SC) once daily. By 12 hours from admission GCS was 15, nasogastric drainage had ceased, her pulse rate was 90–100 bpm, and blood pressure 100–110/60–70 mmHg. Bedside capillary blood glucose was consistently in the range 5–8 mmol/L (90–145 mg/dL), venous pH was 7.32, and creatinine normal.
The first dose of basal insulin was then administered while the nasogastric tube and urinary catheter were withdrawn. Intravenous (IV) fluids and insulin were continued for a further 24 hours alongside restitution of basal bolus SC insulin and normal eating until the urine was free of ketones.
During the period in which the mother was being resuscitated, fetal movements and heart rate were monitored intensively. An ultrasound scan showed the head circumference to be on the 50th centile and abdominal circumference on the 95th centile. There were occasional heart rate decelerations but none was sustained. On day 3 after admission it was planned to administer betamethasone to induce fetal surfactant. However, before this was given, fetal movements ceased and severe decelerations were suddenly recorded. An emergency lower segment cesarean section was performed but the baby was dead on delivery.
- What clinical symptoms and signs suggested DKA?
- What biochemical fi ndings confirmed DKA?
- Why did DKA develop?
- Could DKA have been prevented?
- Why was a nasogastric tube inserted?
- Why was tinzaparin given?
- Why was there a leukocytosis?
- Why did the baby die?
BACKGROUND
DKA is one of a very small number of critical illnesses that occur with any regularity in pregnant women. However, although the frequency of pregnancy in women with diabetes preceding pregnancy is increasing, it is still uncommon and a large portion of the increase is due to the rise of Type 2 diabetes in women of reproductive age. Historically, DKA has been reported to occur in 1–3% of women with diabetes preceding pregnancy,1,2 but these studies were in an era when almost all diabetes preceding pregnancy was Type 1 and insulin management was less intensive. So, since treatment has become more intensive, and as DKA occurs much more rarely in Type 2 diabetes than in Type 1 diabetes (particularly when diabetes duration is short), it is probable that the incidence is now much lower.
DKA occurs when profound insulin deficiency is combined with increased catabolic hormone concentrations. In consequence there is over production from the liver of both glucose and ketone bodies. This over production then combines with progressively decreasing tissue and renal clearance of glucose and ketoacids to produce an accelerating spiral of worsening hyperglycemia, hyperketonemia, and acidosis, the classical triad of DKA. 3–5
ETIOLOGY
Infection is a classical precipitant of DKA because it induces a release of catabolic hormones such as epinephrine, cortisol, glucagon, and growth hormone. More common causes, however, include management errors, omission of insulin, and new cases of undiagnosed diabetes. Because DKA occurs only in the context of severe insulin deficiency, the majority of patients have Type 1 diabetes. However, ketoacidosis can sometimes be precipitated in Type 2 diabetes. Furthermore subtypes of Type 2 diabetes occur in certain ethnic groups in whom the development of ketoacidosis seems to occur more readily.
The sequence of metabolic changes leading to ketoacidosis is depicted in Fig. 19.1. In the early stages of DKA, the profound insulin deficiency leads not only to hyperglycemia but also to a rise in plasma glucagon which in turn stimulates hepatic gluconeogenesis and lipolysis. Hyperglycemia and ketoacidosis then cause osmotic diuresis, vomiting, hyperventilation, and vasodilatation, leading to hypotension which stimulates the release of catecholamines and cortisol, in turn leading to a vicious cycle of worsening metabolic decompensation provocative of further metabolic hormone release. Simultaneously, insulin deficiency reduces insulin-stimulated glucose disposal in peripheral tissues, such as muscle and adipose tissue, thereby fuelling the rise in glucose.
The combination of severe insulin deficiency and excess catabolic hormones promotes the breakdown of adipose tissue triglyceride (lipolysis), leading to the release of large quantities of long chain non-esterified fatty acids (NEFAs) into the circulation. These are the principal substrate for ketogenesis in the liver. Insulin deficiency and high catabolic hormone levels also promote ketogenesis. Because ketone bodies are strong organic acids that dissociate fully at physiologic pH, the increased levels rapidly outstrip the buffering capacity of the body fluids and tissues, leading to metabolic acidosis.
Hyperglycemia and acidosis both contribute to fluid and electrolyte depletion. Hyperglycemia causes an osmotic diuresis and this is compounded by ketonuria, leading to substantial loss of water and electrolytes (dehydration). Insulin deficiency and glucagon excess exacerbate sodium loss in the kidney. Metabolic acidosis leads to exchange of intracellular potassium ions for extracellular hydrogen ions, the potassium then being lost in excreted body fluids. The body attempts to compensate for the excessive metabolic acids by hyperventilation.
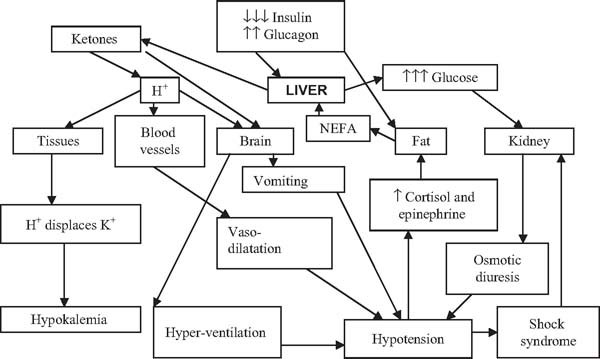
Fig. 19.1 Pathogenesis of diabetic ketoacidosis. NEFAs, non-essential fatty acids.
Further loss of electrolytes occurs because of acidosis-induced vomiting. The loss of body water in ketoacidotic adults is often 5 L or more. When dehydration progresses to the point of hypovolemic shock, reducing renal blood flow inhibits elimination of glucose and ketone bodies, so fuelling rises in their plasma concentrations. Phosphate and magnesium deficiency commonly accompany ketoacidosis but whether they are of clinical significance is uncertain.
Thus the patient with ketoacidosis has:
- Profound insulin deficiency
- High levels of catabolic hormones (glucagon, cortisol, epinephrine)
- Severe hyperglycemia
- Significant acidosis
- Dehydration
- Potassium depletion.
These are the key metabolic derangements that underpin the clinical presentation, natural history, and treatment rationale.
PREGNANCY AS A RISK FACTOR FOR DIABETIC KETOACIDOSIS
“Facilitated a nabolism” and “a ccelerated s tarvation”
The metabolic characteristics of pregnancy may predispose to the development of ketoacidosis. One important influence is human placental lactogen (hPL), a single chain peptide hormone synthesized by the trophoblast and released into the maternal blood. This hormone has the principal action of increasing the supply of glucose to the fetus by decreasing maternal stores of fatty acids. It does this by increasing maternal secretion of insulin. hPL also induces a reduction in maternal insulin sensitivity. Thus during pregnancy, maternal postprandial glucose concentration is greater than that in the non-pregnant state. This in turn increases the gradient in glucose concentration between mother and fetus, enhancing glucose transfer from the former to the latter. In the fasting state, enhanced insulin resistance at the level of the fat cells promotes the release of non-essential fatty acids (NEFAs) for use as a maternal alternative fuel, thereby sparing maternal glucose for fetal utilization. These processes of “facilitated anabolism”6 and “accelerated starvation”7 make women with pregestational diabetes more susceptible to diabetic ketoacidosis which, when initiated, can develop quickly and sometimes at relatively low levels of hyperglycemia.1
Continuous subcutaneous insulin infusion (or “pump” treatment)
The use of continuous SC insulin infusion (CSII) is popular in pregnancy and because of the very small insulin reservoir, this may also represent a risk factor, although the increase in ketoacidosis found in early studies of CSII has been less evident in recent publications8,9 (see chapter 10). In addition Type 1 diabetes, for which DKA is a well-recognized presenting syndrome, is more likely to present during pregnancy because of the prevailing metabolic milieu.
High-dose glucocorticoids
Glucocorticoids are commonly used in high doses to induce fetal surfactant during pregnancies where there is a high risk of impending preterm delivery. In women with diabetes during pregnancy, high-dose glucocorticoids inevitably induce severe hyperglycemia10 unless prompt compensatory management is coadministered.11 This is therefore a situation in which very careful monitoring for the potential development of ketoacidosis is mandatory (see chapter 21).
Tocolytics
Similarly, beta-agonist medications, such as ritodrine and terbutaline, may sometimes be used to try and halt the onset of preterm labour. These drugs also can induce severe hyperglycemia in the context of diabetes during pregnancy and similar vigilance must be exercised to that recommended during the use of glucocorticoids. The combined use of glucocorticoids and beta-agonists is especially hazardous (see chapter 21).
RISKS TO MOTHER AND FETUS OF DIABETIC KETOACIDOSIS
Mother
DKA continues overall to carry an appreciable mortality (around 5% in Western countries), although fatal outcome tends to be concentrated at the extremes of age. Pregnant women who develop DKA may die from the same causes as their non-pregnant counterparts, and there are certain aspects of pregnancy that may render them more vulnerable.
The five major causes of death directly attributable to ketoacidosis (as opposed to a precipitating illness) are:
- Hypovolemic shock
- Aspiration pneumonia
- Cerebral edema
- Hypokalemia induced cardiac dysrhythmias
- Pulmonary embolism.
Hypovolemic shock
This is due to loss of fluid from osmotic diuresis and vomiting, possibly compounded by acidosis-induced hyperventilation and vasodilatation. It will respond to fluid replacement. Rarely acute tubular necrosis may lead to the need for temporary renal replacement therapy.
Aspiration pneumonia
This is a risk predominantly in those patients with ketoacidosis who have impairment of consciousness. Hyperglycemia inhibits gastric emptying and is a major contributory factor in the severity of DKA-associated vomiting. Pregnancy itself, of course, may be associated with decreased gastric emptying and a propensity to vomiting, and therefore may compound this component of the syndrome.
Cerebral edema
Cerebral edema occurs predominately in the young and the old, and especially in females. Thus, pregnant teenagers with Type 1 diabetes who develop DKA are at particularly high risk. The pathogenesis is thought to involve delayed normalization of the intracerebral defenses against dehydration (generation of intraneuronal osmotically active molecules that counterbalance the extra- to intra-cellular osmotic gradient). Over-rapid rehydration during treatment for DKA may contribute to the chance of cerebral edema. Accordingly, it is believed that the risk can be mitigated by avoiding over-rapid rehydration. So, once an adequate circulating volume has been re-established, the reduction in plasma osmolality should proceed gradually, giving the physiologic defenses time to readjust. Typically such patients appear initially to improve as their circulating volume is restored, and then they exhibit a secondary decline in cerebral function as cerebral edema develops.
Hypokalemia
Hypokalemia is an important risk factor for cardiac dysrhythmia, particularly if its onset is rapid. All patients with DKA have total body potassium depletion but because of potassium–hydrogen ion exchange and impaired renal function, the plasma potassium may be normal or even high at presentation. Hypokalemia generally occurs when potassium replacement is not started sufficiently early or in sufficient quantity to replace intracellular potassium, as it normalizes rapidly during metabolic management.
Pulmonary embolism
Patients with ketoacidosis are at increased risk of thromboembolism; the greater the hyperosmolality (hemoconcentration) the greater the risk. Pregnant women are already at increased risk of thromboembolism and therefore this is another component of the ketoacidosis syndrome which is potentially compounded by pregnancy. Prophylaxis with heparin is indicated.
Fetus
Fetal distress and intrauterine death may be associated with ketoacidosis.12 Furthermore, there is some evidence that ketonemia during pregnancy has an adverse impact on the behavioral and intellectual development of the fetus.13 DKA is therefore a very high-risk complication for fetal health.
The causes of fetal loss are thought to be:
- Reduced uteroplacental blood flow (osmotic diuresis/maternal acidosis)
- Maternal acidosis leading to fetal acidosis and electrolyte imbalance
- Maternal hypokalemia leading to fetal hypokalemia with myocardial suppression, and fetal arrthymia
- Maternal hypophosphatemia leading to reduced 2,3-diphosphoglycerate (2,3-DPG)
- Fetal hyperinsulinemia leading to increased fetal oxygen requirement.
CLINICAL MANAGEMENT
Clinical presentation
DKA usually evolves rapidly, i.e. in less than 24 hours. Typically the osmotic diuresis causes severe polyuria and polydipsia. As the degree and duration of hyperglycemia and dehydration progress, lethargy, confusion, and coma may supervene. Acidosis drives the development of vomiting, abdominal pain, and hyperventilation. There may be coexistent symptoms of intercurrent infections, such as productive cough and pleurisy in pneumonia, dysuria and loin pain in pyelonephritis, and fever and rigors in either.
The clinical signs are related to acidosis (hyperventilation, peripheral vasodilatation), hypovolemia (tachycardia, hypotension), and hyperglycemia (succussion splash due to delayed gastric emptying, coma due to cerebral dehydration). There may also be signs of intercurrent precipitating illnesses, such as pneumonia or pyelonephritis. Fever is rare even in the presence of infection because of peripheral vasodilatation, and indeed hypothermia is a potential complication.
The patient will usually report a recent history, including polyuria and polydipsia (and polyphagia?), possibly accompanied by rapid weight loss and generalized weakness. However, history taking may be compromised by discomfort due to abdominal pain, vomiting, drowsiness, confusion or even coma. Difficulty in diagnosis may be encountered in that polyuria, nausea, and abdominal cramping may be normal findings during pregnancy.
Assessment
Physiologic assessment
Hypotension and tachycardia, the cardinal signs of hypvolemia due to dehydration, will be prominent in severe cases; less severe presentations may exhibit only postural hypotension. Metabolic acidosis stimulates the medullary respiratory centre. The resulting breathing pattern is of rapid and deep respirations, sometimes called “air hunger” (Kussmaul respiration). Those who are able to detect it may smell acetone in the breath.
Conscious level must be carefully charted because, as indicated above, any secondary decline in conscious level during treatment must always suggest the possibility of cerebral edema.
Despite hypovolemia, which would ordinarily induce peripheral vasoconstriction, patients will usually be vasodilated because of the acidosis. This may in certain circumstances lead to hypothermia or to the masking of pyrexia. Pregnancy itself is a state of physiologic vasodilatation. Generalized abdominal discomfort and gastric splash are not unusual.
Bedside biochemical measures can usually confirm the diagnosis before laboratory blood results are available. Thus, in the context of the clinical features described above, bedside tests indicating hyperglycemia and heavy (+++) ketonuria or ketonemia are essentially confirmatory of ketoacidosis.
Laboratory assessment
DKA is characterized by the triad of hyperglycemia, ketonemia, and anion gap metabolic acidosis.
Hyperglycemia
The serum glucose concentration is usually greater than 25 mmol/L (456 mg/dL). Because of fasting insulin resistance during pregnancy, ketoacids form at lower maternal glucose concentrations than in the non-pregnant state. Therefore, DKA may develop at glucose concentrations less than 25 mmol/L (456 mg/dL)14.
Ketonemia
Three ketone bodies are produced in DKA: two ketoacids (β-hydroxybutyric acid and acetoacetic acid) and one neutral ketone (acetone). Testing for ketones can be carried out on urine or serum with nitroprusside-based reagent sticks. Nitroprusside reacts with acetoacetate and acetone but not with β–hydroxybutyrate, so that very occasionally a negative nitroprusside reaction is obtained in the presence of severe ketosis. A bedside capillary measure of β-hydroxybutyrate is routinely employed in some centers and has the advantage of providing an easily obtainable numerical index of successful management.15
Metabolic acidosis
The anion gap metabolic acidosis is due to the production and accumulation of β-hydroxybutyric acid and acetoacetic acid. Compensatory hyperventilation leads to loss of CO2 and reduction of serum bicarbonate concentration, thus attenuating the fall in arterial pH. Typically in ketoacidosis, pH is between 6.9 and 7.3. Physiologically the serum anion gap provides an estimate of unmeasured anions in the serum, such as albumin. In metabolic acidosis it is increased by the presence of the pathologic anions, i.e. in DKA ketoacids. The anion gap is calculated by subtracting the major measured anions, chloride and bicarbonate, from the major measured cation sodium. Physiologically the gap is less that 15 mmol/L (<273 mg/dL). Patients with DKA usually have an anion gap of greater than 20 mmol//L (20 mEq/L). The normal anion gap of about 10–15 mmol/L (10–15 mEq/L) is accounted for by phosphate, sulfate and lactate ions, and ionized proteins. In the context of hyperglycemia and detectable ketones, none of the other causes of anion gap acidosis is likely (alcoholic ketoacidosis, lactic acidosis, advanced endstage chronic renal failure or poisoning [with salicylates, ethylene glycol or methanol]).
Other laboratory measurements
Sodium
Serum sodium concentration in ketoacidosis is variable. Osmotic water movement out of the cells, driven by hyperglycemia, leads to dilution and a reduction in sodium concentration, whereas glycosuria-induced osmotic diuresis results in water loss in excess of sodium and an increase in the sodium level. Occasionally the measured sodium concentration may be misleadingly low as a result of the phenomenon of pseudohyponatremia, in which DKA-associated hyperlipidemia is so severe that the serum is milky and contains less water and therefore less sodium.
Potassium
On presentation patients with DKA have an average potassium deficit of 3–5 mmol/kg. This is due to a combination of the cellular transmembrane exchange of potassium for hydrogen ions, the renal potassium losses accompanying the glucose-induced osmotic diuresis, and gastrointestinal losses due to vomiting. However, initially, because of displacement of intracellular potassium, dehydration-related hemoconcentration, and hypovo-lemic renal impairment, potassium levels in the serum are commonly normal, disguising the underlying whole body deficiency.
Urea and creatinine
When available, the initial laboratory tests typically show elevated urea and creatinine in keeping with acute renal impairment. In the past, plasma creatinine concentrations may have been falsely elevated in ketoacidosis due to assay interference by acetoacetate but this is now unusual.
Blood count
The majority of patients with ketoacidosis have a leuko-cytosis but this does not necessarily imply associated infection. Hemoglobin may initially be high due to dehydration-related hemoconcentration.
Other laboratory measurements
A variety of enzymes, including amylase, transaminases and creatinine kinase, may be raised non-specifically. Significant hyperlipidemia, hypophosphatemia, and hypomagnesemia may also occur.
Other tests
If there is a suggestive history or clinical signs, chest X-ray may be appropriate to confirm pneumonia, urine culture to look for urinary infection or blood culture to diagnose septicemia.
TREATMENT
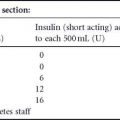
Two protocols for the management of DKA from different UK centers are given by way of illustration (Tables 19.1 and 19.2). Each of the protocols is broadly similar and they generally concur with American Diabetes Association (ADA) recommendations. The first used by the author involves the infusion of insulin at a dose dictated by the ambient glucose and total daily insulin dose preceding DKA. The second protocol employed by the Belfast group includes a preceding insulin bolus followed by an extended insulin regimen, and utilizes capillary ketone body measurements both in the management and as a point of titration for the hourly insulin infusion. General principles of management are similar and need to be followed rigorously.
Table 19.1 Protocol 1: Management of diabetic ketoacidosis.