- 10.1 Introduction
- 10.2 Patterns of joint disease
- 10.3 Arthritis and infection
- 10.3.1 Septic arthritis
- 10.4 Rheumatoid arthritis [RA]
- 10.4.1 Diagnosis
- 10.4.2 Serology
- 10.4.3 Pathology
- 10.4.4 Immunopathogenesis
- 10.4.5 Aetiology
- 10.4.6 Outcome
- 10.4.7 Management
- 10.4.1 Diagnosis
- 10.5 Seronegative spondyloarthritis
- 10.5.1 Ankylosing spondylitis
- 10.5.2 Other seronegative spondyloarthritides
- 10.5.3 Other seronegative arthritides
- 10.5.1 Ankylosing spondylitis
- 10.6 Chronic arthritis in children
- 10.7 Systemic lupus erythematosus
- 10.7.1 Clinical features
- 10.7.2 Laboratory findings
- 10.7.3 Differential diagnosis
- 10.7.4 Drug-induced systemic lupus erythematosus
- 10.7.5 Management of systemic lupus erythematosus
- 10.7.6 Prognosis
- 10.7.7 Aetiology and pathogenesis
- 10.7.1 Clinical features
- 10.8 Other ‘connective tissue’ diseases
- 10.8.1 Mixed connective tissue disease
- 10.8.2 Sjögren’s syndrome
- 10.8.3 Scleroderma
- 10.8.1 Mixed connective tissue disease
- 10.9 Systemic vasculitis
- 10.9.1 Polyarteritis nodosa
- 10.9.2 Polymyalgia rheumatica and giant cell (temporal) arteritis
- 10.9.3 Other vasculitides
- 10.9.1 Polyarteritis nodosa
- 10.10 Inflammatory muscle disease or myositis
- 10.11 Hereditary periodic fevers
 Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
10.1 Introduction
Immunological mechanisms are responsible for many rheumatological diseases. Although these disorders often present with joint or muscle inflammation, many have multisystem involvement with a particular tendency to involve skin, lungs and kidney. These disorders are often autoimmune and include rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), dermatomyositis, scleroderma and some forms of vasculitis. They used to be referred to as the ‘connective tissue diseases’; this woolly phrase was meaningless in pathophysiological terms, and the phrase largely dropped from usage. Discussion of these disorders can also be found in other organ-based chapters in relation to the particular organ involved. These systemic autoimmune diseases are characteristically associated with non-organ-specific autoantibody production, particularly against components of cell nuclei. These autoantibodies are not necessarily responsible for joint or tissue damage, but are helpful markers in diagnosis or prognosis (see Chapter 19).
10.2 Patterns of joint disease
Joint diseases fall into two broad categories: degenerative conditions of cartilage (osteoarthritis) or disorders characterized by inflammation of the joint lining or synovium (inflammatory arthritis or synovitis). The diagnosis of a particular form of inflammatory arthritis is rarely made using a single diagnostic test, notable exceptions being the presence in synovial fluid of uric acid crystals in gout or of bacteria in septic arthritis. Instead, the diagnostic process relies heavily upon clinical assessment, with a smaller role being played by various immunological tests (Table 10.1). Most forms of chronic inflammatory joint diseases can be defined using clinical criteria alone, and this, in part, reflects our ignorance of the underlying aetiopathogenesis. The major common patterns of joint inflammation and the common underlying causes are summarized in Table 10.2.
Table 10.1 Major factors in the assessment of the patient with joint disease
| Age/gender |
| Acute or chronic |
Pattern of joint disease
|
| Extra-articular disease |
| Rheumatoid factor positive |
| Antinuclear antibody positive |
Table 10.2 Common patterns of inflammatory joint disease
| Pattern of joint inflammation | Common causes |
|---|---|
| Monoarthritis | Bacterial infection |
| Gout/pseudogout | |
| Spondyloarthropathy (especially reactive arthritis) | |
| Oligoarthritis | Spondyloarthritis (especially reactive and psoriatic) |
| Gout/pseudogout | |
| Polyarthritis | |
| Symmetrical | Rheumatoid arthritis |
| Systemic lupus erythematosus | |
| Viral arthritis | |
| Asymmetrical | Psoriatic arthritis |
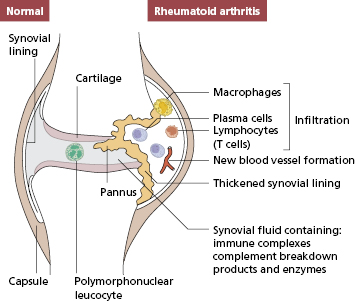
The healthy synovial lining (Fig. 10.1) consists of a single layer of cells overlying loose, well-vascularized stromal tissue. The lining cells are of two kinds: one fibroblastic, which synthesizes the proteoglycans, which act as a lubricant within the joint, and one derived from macrophages, which probably have a scavenging function. Unlike most body surfaces, free passage of intercellular fluid can occur across the synovial membrane, a factor that may explain why antigens tend to be deposited within the joint. The synovial response to injury is relatively limited, and the pathology of most forms of inflammatory arthritis consists of hyperplasia of the lining layer and cellular infiltration of the vascular tissues beneath the lining. Cytokine production in chronic synovial inflammation may also be limited in its variety: most synovial diseases show some response to treatments that block the action of tumour necrosis factor (TNF)-α.
Fig. 10.1 Diagrammatic representation of a joint. One side is normal, the other shows characteristic pathological features of rheumatoid arthritis.
10.3 Arthritis and infection
There are two principal mechanisms whereby microorganisms can cause inflammatory arthritis: direct invasion of the synovium (infective or septic arthritis) or a hypersensitivity response (mediated by T-cell or immune complexes) against microbial antigen deposited within the joint.
10.3.1 Septic arthritis
Pyogenic arthritis can occur in previously healthy joints in immunocompetent subjects, but the risk is greatly increased by previous joint damage or defective immunity, particularly abnormalities of antibody production or neutrophils (the latter being most often a consequence of chemotherapy). Joint damage probably increases the risk of sepsis by allowing increased entry of organisms into the joint. Patients with RA have a particular propensity to joint sepsis, this being partly due to corticosteroid therapy and joint damage, but also related to subtle defects in immunity associated with the disease itself (Case 10.1). The organisms most frequently associated with septic arthritis are summarized in Table 10.3.
 Case 10.1 Septic arthritis
Case 10.1 Septic arthritisA 37-year-old woman developed a symmetrical polyarthritis. A test for CCP antibodies was positive and erosive changes were seen on X-ray, confirming a clinical diagnosis of RA. The arthritis followed an aggressive course with poor response to a variety of disease-modifying antirheumatic drugs and she became increasingly disabled due to severe destructive changes in the knees, wrists and shoulders. A moderate dose of prednisolone was introduced at the age of 42, with some symptomatic improvement in her joints and she was referred to an orthopaedic surgeon with a view to knee replacements. However, 1 month before her orthopaedic appointment she presented to the emergency department with a painful swollen right knee. On examination she was unwell, febrile (38 °C) and had a hot, red right knee with a sizable effusion. Eighty millilitres of purulent synovial fluid was aspirated from the joint and microscopy of the fluid revealed numerous Gram-positive cocci. A diagnosis of septic arthritis was made on a background of severe RA and steroid therapy. She was treated with high-dose IV antibiotics and the joint was washed out via an arthroscope. Culture of blood and synovial fluid grew Staphylococcus aureus. She received 6 weeks’ antibiotic treatment in total together with vigorous physiotherapy. Her knee, however, was significantly worsened by the infection and she could no longer straighten the leg or walk more than a few yards. Joint replacement was deferred for 6 months to reduce the risk of infection in the prosthesis.
Table 10.3 Relative frequency of bacterial causes of septic arthritis in the UK
| Staphylococcus aureus | 40% |
| Pneumococcus | 10% |
| Other streptococci | 18% |
| Haemophilus influenzae | 7% |
| Gram-negative bacilli | 12% |
| Gonococcus* | <1% |
*Gonococcal joint infection is much more common in North America and Australia.
Septic arthritis is a medical emergency. Delay in treatment is associated with an increasing risk of severe joint damage and with high mortality in immunocompromised subjects.
Gonococcal and meningococcal infection can be associated with arthritis. This is most often associated with a subacute septicaemic illness, and organisms can be isolated from the blood and synovial fluid. However, in gonococcal (and less commonly meningococcal) infection, an immune complex-mediated arthropathy can also occur, which usually presents 7–10 days after the onset of infection and is associated with falling levels of antigen in serum, rising levels of antibody and evidence of complement consumption, all features suggesting an immune complex-mediated disease.
Lyme disease, which is caused by the tick-borne spirochaete Borrelia burgdorferi, is associated with a chronic large-joint arthritis. Borrelia are invasive, non-toxigenic, persistent pathogens, and little is known about the pathogenesis of Lyme disease. The arthritis first appears some months after the initial tick bite, and usually has a relapsing and remitting course, but can be persistent and associated with joint destruction. Organisms are difficult to isolate from the joint, but an antibody response can be detected. The arthritis may be partly mediated by hypersensitivity mechanisms but improves after antibiotic treatment, suggesting that live organisms also play a role in pathogenesis.
Viral arthritis
Joint and muscle pain is very common in acute viral infections, but inflammatory arthritis is much less common. Infections such as rubella, mumps and hepatitis B can cause a transient arthritis that is probably due to a combination of direct infection and hypersensitivity, since immunization with attenuated rubella virus can also cause a transient arthritis. The most common viral cause of arthritis in adults is parvovirus B19 (which in children usually only produces a mild febrile illness with a characteristic ‘slapped cheek’ rash). This produces a symmetrical, peripheral polyarthritis (clinically similar to early RA), which usually remits within 2 weeks but which can occasionally persist for several months.
Immune-mediated arthritides
The distinction between active infection and immune-mediated arthritis is not always clear-cut, as the earlier account makes clear. There are other forms of arthritis that appear to be triggered by infection and which follow a subacute, relapsing or chronic course. The most common of these, reactive arthritis, is discussed further in section 10.5. It will also be apparent from the sections on RA, other spondyloarthritides and juvenile chronic arthritis that attempts have been made to explain most forms of chronic inflammatory joint disease in terms of inappropriate response to an unidentified organism. This model has perhaps been most successfully applied to the spondyloarthritides (see section 10.5). Other disorders in which arthritis is immune mediated include rheumatic fever, which is discussed in Chapter 2, and Henoch–Schönlein purpura, discussed in Chapter 9.
10.4 Rheumatoid arthritis [RA]
10.4.1 Diagnosis
RA is a common disease affecting 1–2% of the adult population and is twice as common in women as men. It is most common between the ages of 25 and 55 years, and the most frequent presentation is an insidious, symmetrical polyarthritis, although the disease can begin abruptly. Although systemic manifestations (Table 10.4) may be present at the outset, they develop more usually as the disease progresses (see Case 10.3) The diagnosis of RA is made on clinical grounds, as illustrated in Case 10.2.
Table 10.4 Extra-articular features of rheumatoid arthritis: the commonest features are marked in bold
Intrinsic or essential systemic features of rheumatoid disease Rheumatoid nodules Serositis (pleurisy, pericarditis) Vasculitis Neuropathy (often caused by vasculitic damage to nerves) Felty’s syndrome (neutropenia and splenomegaly) |
Consequences of chronic inflammation and immune stimulation Anaemia Weight loss and fever Lymphadenopathy |
Complications of rheumatoid disease Entrapment neuropathy, e.g. carpal tunnel syndrome Accelerated atherosclerosis Infection Amyloidosis Cervical myelopathy |
Complications of treatment Osteoporosis (corticosteroids) Accelerated atherosclerosis Infection Drug-specific side-effects |
Associated syndromes (also occur without RA) Keratoconjunctivitis sicca Interstitial lung diseases Episcleritis and scleritis |
 Case 10.2 Rheumatoid arthritis – classic course
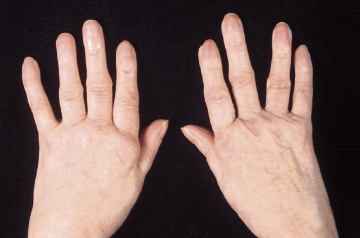
Case 10.2 Rheumatoid arthritis – classic courseA 37-year-old woman gradually developed painful wrists over 3 months; she consulted her doctor only when the pain and early morning stiffness stopped her from gardening. On examination, both wrists and the metacarpophalangeal joints of both hands were swollen and tender but not deformed (Fig. 10.2). There were no nodules or vasculitic lesions. On investigation, she was found to have a raised C-reactive protein (CRP) level (27 mg/l) (NR <10) but a normal haemoglobin and white cell count. A latex test for rheumatoid factor was negative but autoantibodies to cyclic citrullinated peptides were detected.
Fig. 10.2 Symmetrical synovitis in early rheumatoid arthritis. From Medical Masterclass. Rheumatology and Clinical Immunology, 1st Ed. Copyright © 2001 Royal College of Physicians. Reproduced with permission.
The clinical diagnosis was early RA and she was treated with ibuprofen. Despite some initial symptomatic improvement, the pain, stiffness and swelling of the hands persisted and 1 month later both knees became similarly affected. She was referred to a rheumatologist.
She was seen 3 months after the initial presentation. By this time she was struggling to work, drive and carry heavy objects. A test for rheumatoid factor was now positive (titre 1/256) and anti-CCP antibodies were strongly positive. X-rays of the feet showed small but definite erosions in two metatarsal heads. She still had a raised CRP (43 mg/l) but normal serum complement (C3 and C4) levels.
This woman had rheumatoid arthritis with some features suggesting a poor prognosis. Her treatment was changed to weekly low-dose methotrexate. This controlled the arthritis and no further erosions developed for several years. When the disease flared, she received anti-TNF therapy. In view of the poor prognosis, she would now be started on anti-TFN therapy by her rheumatologist when first seen in clinic.
10.4.2 Serology
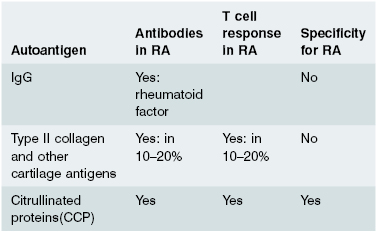
A relatively new test for rheumatoid arthritis measures levels of antibodies that bind citrulline modified proteins (anti-CCP) is now widely available. It is specific for rheumatoid disease as it is elevated in patients with RA or in those about to develop RA and not in other forms of arthritis (see later in the text).
Patients with RA fall into two groups: those with usually less severe disease who lack circulating rheumatoid factor (seronegative RA); and a larger group (70%) with more aggressive disease who have, or develop, a rheumatoid factor in the serum (seropositive RA). Extra-articular disease occurs predominantly in this seropositive group. Rheumatoid factor is only of value in prognosis, not in diagnosis. Rheumatoid factor, in this context, refers only to the IgM antibody which binds aggregated IgG as its antigen; rheumatoid factors of the IgG or IgA class are not clinically helpful, although they may play a role in joint inflammation. The presence of anti-CCP antibodies can also be used to predict which patients will get more severe rheumatoid arthritis. Other serological tests are of little value in RA. Antinuclear antibody (ANA) is present in 40%, but is usually of low titre and often of IgM class; such ANAs are found in many other conditions. Serum complement C3 and C4 levels may be normal or raised due to an ‘acute-phase’ reaction (see Chapter 1).
C-reactive protein (CRP) (another ‘acute-phase’ reactant) is a most important test in any inflammatory arthritis, being raised particularly in active RA; this helps to distinguish active RA from active SLE in which there is not an acute-phase response (see Fig. 10.8). Some hospitals still use an ESR or a plasma viscosity to indicate inflammation but these tests are less reliable and more expensive.
10.4.3 Pathology
The earliest pathological change in RA appears to be an infiltrate of neutrophils and mononuclear cells around small blood vessels in the loose connective tissue beneath the synovium. As the disease progresses, large numbers of T cells, macrophages, B cells and plasma cells accumulate in this tissue, and the synovium becomes markedly thickened due to fibroblast proliferation and macrophage migration. The surface area increases and becomes folded into villi. New vessel formation (angiogenesis) occurs and some blood vessels develop into structures specialized for lymphocyte migration: so-called high endothelial venules (see section 1.7). Secondary lymphoid follicles also develop. In consequence, the histological structure comes to resemble that of an activated lymph node, emphasizing the high degree of immunological activity in RA. Increased amounts of synovial fluid can form (especially in large joints), and this contains large numbers of neutrophil polymorphs, which have migrated from the blood.
This chronic inflammatory tissue has several destructive effects upon the joint. First, the hypertrophied lining layer at the margins of the joint (known as pannus) erodes cartilage and underlying bone. Second, cytokines induce chondrocytes and fibroblasts to produce enzymes that break down the extracellular matrix. Third, degranulation of neutrophils in the synovial fluid can directly damage the surface of cartilage (Fig. 10.1).
10.4.4 Immunopathogenesis
The immunohistology of rheumatoid synovium suggests that the disease results from an immunological response to an unknown antigen within the joint. It is, however, clear that joint destruction in RA is driven by activated macrophages (Fig. 10.3), which secrete TNF-α and other cytokines to cause tissue breakdown directly as well as activating other cells in the joint. A large number of inflammatory mediators are produced in RA synovium (Fig. 10.3), and early descriptions of cytokine production in RA talked of a ‘cytokine soup’, suggesting no simple way of inhibiting this process. However TNF-α plays a central role (Fig. 10.4) since inhibition with neutralizing antibodies reduces the production of other inflammatory mediators in cell culture. Furthermore, when these antibodies are administered to patients, a dramatic improvement in joint inflammation occurs. The members of the interleukin (IL)-1 family, IL-1α and IL-1β, also play an important role, although less crucial than TNF.
Fig. 10.3 How CD4+ T cells and immune complexes may control the events leading to joint damage in rheumatoid arthritis.
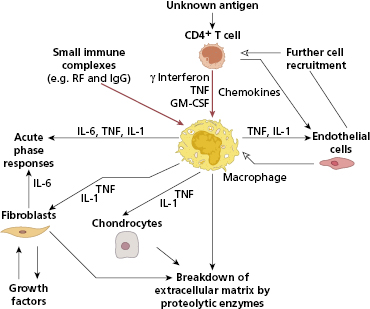
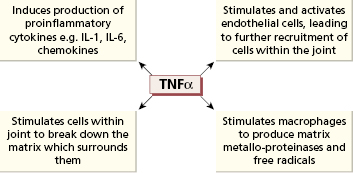
There is probably more than one immunological mechanism leading to macrophage activation in rheumatoid synovium. The most widely accepted model is a process driven by CD4+ Th1 cells. The evidence for T-cell involvement in RA is summarized in Table 10.5. However, the model of RA as a T-cell-driven disease has been criticized, as T-cell cytokines such as IL-2 and interferon (IFN)-γ are hard to detect in RA synovium, in contrast to macrophage-derived cytokines such as TNF-α, IL-1 and IL-6, IL-21 and IL-23, which are present in abundance. Following the descriptions of Th17 cells being central to inflammation and RA being an inflammatory disease par excellence, the role of these cells makes sense given the pathology. Th17 cells form a subset that produces interleukin-17A, 17F, IL-21 and IL-22 as well as TNF-α. Th17 differentiation is promoted by macrophage and DC derived TGF-β as well as the cytokines mentioned –IL-1β, IL-6, IL-21 and IL-23. At the same time Tregs are suppressed, shifting T-cell homeostasis toward inflammation. Th17 cells primarily produce IL-17A, which together with TNF-α activates fibroblasts and chondrocytes. Foxp3+ Tregs, although detected in tissues from patients with RA, have limited function.
Table 10.5 Evidence that T and B cells play a central role in the pathogenesis of rheumatoid arthritis (RA)
| T cells |
|
| B cells |
|
Inflammation driven by antibodies may also be important. The synovium is infiltrated by B cells and may even form ectopic lymphoid follicles. Plasmablasts and plasma cells are widely distributed in the synovium and in juxta-articular bone marrow. Clinical demonstration that depletion of B cells (with an anti-CD20 monoclonal antibody, rituximab – see Chapter 7) dramatically improves the clinical features of RA in a subset of patients, shows that antibody-mediated inflammation does play a crucial role in the disease (Table 10.5) though the elimination of activated B cells that can present autoantigen to more T cells and produce inflammatory cytokines such as IL-6 is important too. The role of innate immunity is uncertain as yet, but the ability of macrophages and neutrophils to be activated directly by triggering factors, through TLRs or intracellular receptors, to produce inflammatory cytokines is likely as the monoclonal antibody, Tocilizumab, an IL-6 receptor inhibitor, has been shown to have excellent clinical effects.
Since RA is an immunologically mediated disease, then it should be possible to define the main antigens driving the T- and B-cell responses in the joint. These autoantigens could be self-molecules expressed in the joint or could be foreign (e.g. bacterial or viral) antigens sequestered in joint tissue. T- and B-cell responses to a number of autoantigens have been described in RA (Table 10.6), but most are not specific for RA and the role of responses against these antigens in the disease is unclear. One of the most plausible autoimmune models of RA would be for the disease to be driven by an immune response against an antigen whose expression is confined to joints, such as type II collagen, the main protein of articular cartilage. However, although immune responses against cartilage and synovial proteins can be found in some patients with RA, these are not found in all patients and are also found in other diseases. Antibodies against a number of peptides containing the unusual amino acid, citrulline, (known as anti-cyclic citrullinated peptide antibodies (CCP antibodies) have recently been found to be highly specific to RA and to predict aggressive and persistent disease. It seems likely that RA is not caused by a single antigen, but rather by a response against a range of ‘citrullinated’ joint specific proteins in genetically susceptible patients. In patients with the ‘shared epitope’ (see section 10.4.5), smoking produces detectable citrullinated proteins in the lungs, mechanistically linking genetic and environmental risk factors.
There is also evidence that macrophage-induced inflammation can become self-sustaining and independent of any triggering immune response: cytokines such as TNF-α produced by macrophages induce further macrophage activation. It is plausible that different mechanisms may predominate at different stages of this chronic disease.
10.4.5 Aetiology
In Europeans there is an association between possession of HLA-DR4 and -DR1 and rheumatoid factor-positive RA. This association becomes stronger with increasing severity of disease, and the presence of extra-articular manifestations (Table 10.4). Once the detailed structure of DR molecules was known at the amino acid level, comparison of different DR alleles associated with RA showed that almost all alleles associated with RA (HLA-DRB1*0101, 0401 and 0404) have a distinctive five amino acid sequence near the peptide-binding region of the DRβ chain; this sequence is known as the ‘shared epitope’. Possession of two copies of the ‘shared epitope’ (one inherited from each parent) is not only associated with a high risk of severe disease (Table 10.7) but also with presence of anti-CCP antibodies. Genome-wide association studies have shown that other genes involved in T-cell activation and the NFκB inflammation pathways are important. Female sex is an important genetic risk factor, and it seems plausible that this risk is mediated hormonally as there is an increased risk of onset of RA in the post-partum period.
Table 10.7 How possession of an HLA-DRB1 allele containing the ‘shared epitope’ influences absolute risk of developing rheumatoid arthritis (RA) in a Caucasian population
| Approximate risk of developing RA | |
|---|---|
| Total population | 1 in 100 |
| Subjects with no copies of shared epitope | 1 in 580 |
| DRB1*0401 | 1 in 35 |
| DRB1*0404 | 1 in 20 |
| DRB1*0101 | 1 in 80 |
| DRB1*0401 and 0404 | 1 in 7 |
The risk is highest in those who inherit a copy of a different DRB1 gene from each of their parents.
Environmental factors predisposing to RA remain poorly understood. Smoking is a relatively weak risk factor, but can increase the risk of RA 20- to 40-fold in genetically susceptible individuals. Many attempts have been made to link RA to infectious agents such as parvovirus, Epstein–Barr virus, mycoplasma and mycobacteria. However, while the idea of RA as a disease either initiated or perpetuated by an immune response against an infection has many attractions, no convincing evidence exists to implicate any known agent (see Chapter 5 for discussion of how infections may trigger autoimmunity).
Many animal models of RA have been developed, centred on both infection and autoimmunity, all with some similarity to the human disease. If nothing else, these models emphasize that the synovial response to injury is limited in scope and most of the aetiological factors are plausible and not mutually exclusive.
10.4.6 Outcome
Although the outcome of RA is variable and a few patients have mild or remitting disease, for most it is a severe and disabling illness. Most (70%) of RA patients have many periods of remission and relapse over decades (polycyclic disease as in Case 10.2) though sustained remission is rarely achieved. Joint damage begins in the early stages of the disease and once established is effectively irreversible. Five years after diagnosis, one-third of patients are unable to work at all and only one-third are still in full-time work. At 10 years, 10–20% of those affected are highly dependent upon others for self-care. Factors predicting a poor prognosis are detailed in Table 10.8.
Table 10.8 Predictors of a poor prognosis in early rheumatoid arthritis
| High titre serum rheumatoid factor |
| Female sex |
| High levels of acute phase proteins (ESR, CRP) |
| High levels of disability |
| Extra-articular disease |
| Radiological evidence of joint damage |
| High titre anti-citrullated peptide (anti-CCP) antibodies |
| Possession of the shared epitope: two copies are worse than one |
ESR, Erythrocyte sedimentation rate; CRP, C-reactive protein.
RA is associated with increased mortality as well as disability. Life expectancy in RA is reduced by approximately 7 years compared with age- and sex-matched controls. In severe, disabling RA, the 5-year mortality is around 50%. The causes of these premature deaths are multiple and often not directly attributable to the arthritis, although some patients die of extra-articular disease such as pulmonary fibrosis (Case 10.3), vasculitis, amyloid (see Chapter 9) or lymphoma and others from the side effects of drug treatment. Patients with RA also show an increased susceptibility to severe or recurrent infection. The largest single contributor to increased mortality in RA is, however, an increased risk of ischaemic heart disease (IHD). Mortality from IHD in RA is increased by approximately two- to threefold, a comparable increase in risk to that seen in diabetes mellitus. Similar increased mortality from IHD has also been described in other chronic inflammatory diseases, particularly SLE. The mechanisms underlying this increased risk are unclear, but conventional risk factors alone (such as smoking, hyperlipidaemia and hypertension) cannot explain the increased risk, and immunosuppressive treatment and disease-specific factors make substantial contributions.
 Case 10.3 Pulmonary fibrosis in rheumatoid arthritis
Case 10.3 Pulmonary fibrosis in rheumatoid arthritisA 61-year-old man, with a 15-year history of seropositive RA, was admitted with increasing shortness of breath, myalgia and weight loss. He had previously smoked 40 cigarettes a day but had never been exposed to coal or silica dust. On examination, he was pale and thin, with generalized muscle tenderness. Widespread crepitations were heard over both lung fields. His joints were tender and he had subluxation of the metacarpophalangeal joints of both hands. There was bilateral cervical and axillary lymph node enlargement but no splenomegaly. Neurological and cardiac examinations were normal.
Investigations showed a raised CRP (81 mg/l) and a normochromic anaemia (Hb 95 g/l) but a normal white cell count. His serum IgG was raised at 44 g/l (NR 7.2–19.0), although IgA and IgM levels were normal. He had a strongly positive rheumatoid factor and anti-CCP antibodies titres. There were no detectable antibodies to DNA or to extractable nuclear antigens (ENA) (see Chapter 19
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


