Method
Measurement
Advantages
Disadvantages
Value in drug development decisions
Fasting hepatic DNL
2–3 days of oral administration of deuterated water (2H2O)
The fraction of palmitate that is newly synthesized in the liver
Outpatient, single blood sample, real world measurement
Repeat measurement variable depending on recent diet
Can be used to assess diet/drug interactions in response to chronic treatment; useful for evaluating nonalcoholic fatty liver disease
Fasting hepatic DNL
13C-acetate infusion
The fraction of palmitate that is newly synthesized in the liver
Single blood measurement, known precursor pool enrichment
Requires overnight intravenous infusion. Repeat measurement variable depending on recent diet
Accurately assesses metabolic status and response to chronic treatment
Fructose-stimulated hepatic DNL
13C-acetate infusion and continuous oral fructose feeding
The fraction of palmitate that is newly synthesized in the liver in response to fructose feeding
Very reproducible; less between subject variability compared to fasting hepatic DNL; known precursor pool enrichment
Requires 24-h inpatient intravenous infusion and repeated fructose dosing
Accurate assessment of acute or chronic inhibition of DNL; particularly useful for evaluation of lipogenesis inhibitors
Adipose DNL and triglyceride synthesis
14–21 days of oral administration of deuterated water (2H2O)
The fraction of palmitate and triglyceride that are newly synthesized in adipose tissue
In vivo assessment of fatty acid and triglyceride synthesis rates in adipose tissue; can be combined with adipocyte cell proliferation measurement
Requires adipose tissue sample
Can evaluate chronic effects on adipose growth/metabolism; particularly valuable for weight loss and insulin-sensitizing agents
Glucose Metabolism
Method | Measurement | Advantages | Disadvantages | Value in drug development decisions | |
|---|---|---|---|---|---|
Fasting endogenous glucose production (infusion) | Infusion of a stable isotope-labeled glucose | Rate of endogenous glucose production into plasma | Gold standard, quantifies hepatic and renal contribution to fasting glucose | 5–7-h IV required | Demonstrate effects on regulation of fasting glucose |
Suppression of endogenous glucose production (clamp) | Infusion of a stable isotope-labeled glucose during a hyperinsulinaemic euglycaemic clamp | Rate of endogenous glucose production into plasma | Gold standard for the measurement of hepatic insulin sensitivity, simultaneous measurement of peripheral insulin sensitivity | IV and glucose clamp required, prior knowledge of endogenous glucose production rates needed for optimal infusion protocol design | Evaluation of PK/PD effects on hepatic and peripheral insulin sensitivity |
Fasting endogenous glucose production (bolus) | Intravenous bolus of a stable isotope-labeled glucose with frequent blood sampling | Rate of endogenous glucose production into plasma | Easy to perform, shorter duration than intravenous method | Less validated, data modelling required, frequent blood sampling | Demonstrate effects on regulation of fasting glucose |
Glycolysis | Oral administration of [6,6-2H2] glucose as part of a mixed meal of an oral glucose tolerance test (OGTT) | Disposal of oral glucose through glycolytic pathway (vs. glycogen storage) | Simple oral test to assess glycolysis | Accurate insulin measurements required to properly interpret | Adds additional information about the metabolic fate of glucose in an OGTT or mixed meal |
Postprandial glucose fluxes | Dual tracer approach | Glucose absorption, glucose disposal an endogenous glucose production during a mixed meal | Simplified version of the triple tracer; can be combined with glycolysis measurement | IV and frequent blood sampling required, calculation of parameters based on some assumptions | Useful for evaluating PK/PD effects of glucose absorption inhibitors |
Gluconeogenesis/glycogenolysis | Infusion of 13C-glycerol in combination with mass isotopomer distribution analysis (MIDA) | Contribution of gluconeogenesis vs. glycogenolysis to endogenous glucose production | Can be simultaneously assessed with endogenous glucose production | Glycogenolysis is calculated and not directly measured, MIDA requires highly accurate measurements | Can determine hepatic pathways contributing to hepatic glucose output |
Lipoprotein and Protein Metabolism
Method | Measurement | Advantages | Disadvantages | Value in drug development decisions | |
|---|---|---|---|---|---|
Intravenous bolus | Intravenous bolus of 13C- leucine, multi-compartment modelling | Lipoproteins and protein synthesis rates | Gold standard, Simple administration of tracer, many model parameters accessible | Frequent blood sampling and multi-compartment modelling required | Can determine numerous mechanisms of altering lipoprotein concentrations |
Primed constant infusion | Constant infusion of 2H or 13C labeled leucine and simple precursor/product model | Lipoproteins and protein synthesis rates | Fewer samples required, straightforward modelling | Simple model, ignores some parameters | Can determine mechanisms of altering lipoprotein concentrations and turnover of regulatory proteins |
Deuterated water | Oral administration of 2H2O | Tissue and whole-body protein synthesis rates | Simple oral tracer administration; simple lipoprotein and protein kinetics can be determined from a single blood draw or finger prick | Less useful for careful assessment of rapidly turning over proteins | Suitable for large trials, can concurrently assess lipid and carbohydrate metabolism with same tracer |
Conclusions
Stable isotope tracer methods offer the early clinical investigator unique quantitative tools to evaluate pharmacodynamics in small clinical studies. These methods can also be performed in preclinical studies and provide early proof of mechanism and dose response in humans by demonstrating direct modulation of pathways in cardio-metabolic disease.
Introduction
Metabolic flux refers to transformations of chemicals in living systems in the dimension of time. These transformations may be biochemical in nature and involve synthesis, degradation, intermediary metabolism, storage, or other processes, or they may be spatial in nature and involve processes such as transport, uptake, or secretion. A central element to the definition of metabolic flux is that it is best described quantitatively in terms of the rate (mass/per unit time) at which the processes occur. Careful regulation of the flux through metabolic pathways may be seen as the unifying theme governing all cellular regulatory machinery, the central defining feature of a cell type, and the basis of phenotypic variability among cells and organisms.
There are few fields where measurements of metabolic fluxes have been more effectively utilized than in the study of cardio-metabolic disease. From the elucidation of carbon fluxes in the Krebs cycle [1] to the effect of diet on protein and lipid synthesis [2, 3], isotopes, both radioactive and stable, have been essential in understanding the source and fate of metabolites in cell biology and physiology. With the development of modern mass spectrometric methods, there has been an explosion in the number of pathways which can be accurately measured in vivo using stable isotope labeling techniques. Dozens of analytical methods and clinical protocols can be found in the literature dating back more than 70 years [4].
Regardless of the specific methodology used, the in vivo measurement of fluxes is a unique and powerful approach to assess disease physiology and clinical responses. The measurement of flux rates provides information about what is new within a biological system and how rapidly molecules are being synthesized and degraded. It is useful to see the flux of molecules as consisting of the production (rate of entry) and removal (rate of exit) of a molecule into a compartment in the body, such as the cytosol of a tissue or the bloodstream. Static measurements, in contrast to flux measurements, may provide information on the concentration, content, or structure of components of a system in a compartment in the body, which in turn reflects the balance between the input and output rates. When the input and output rates are the same, the system is at steady state, i.e., there are no changes in concentrations of molecules in the system over the time period studied. However, a system at steady state can have either high flux rates (large numbers of new molecules entering and replacing old molecules) or low flux rates (a small number of new molecules entering and replacing old molecules). Static measurements cannot reveal the turnover within the dynamic system that is at steady state and differentiate between these very different states. Understanding the dynamic state within a biological system and the effect of interventions on that state is especially important in early clinical trials where the duration of intervention is often not sufficient to reach a new steady-state condition as, for example, when a treatment targets a slow turnover molecular process, such as tissue fibrosis and brain myelin content. It is axiomatic that changes in flux necessarily precede changes in concentration and is often of a greater magnitude as counter regulatory pathways will typically feedback and “defend” steady-state concentrations [5, 6].
For the evaluation of early clinical responses to novel therapies, it is important that methods be relatively simple in order to be minimally burdensome on the subjects as well as the clinical research team. Here we present selected applications for the in vivo assessment of lipid, glucose, and protein metabolic flux using stable isotope tracers. The focus will be on methods that have been used effectively in early clinical research studies to study disease pathology and drug efficacy. This is by no means intended to be a review of all the available methods for measuring metabolic fluxes in vivo. Many of the methods described here may be performed with single quadrupole gas chromatography/mass spectroscopy (GC/MS) analysis methods, such as the Agilent 6890’s series instruments which are inexpensive, widely available, and provide excellent quantitative isotope enrichment measurements when carried out properly. In principle, however, any instrument that can accurately measure the relative isotopomer abundance for a specific analyte can be used. Methods such as GC/MS, isotope ratio mass spectrometry (IR/MS), liquid chromatography-tandem mass spectrometry (LC-MS/MS), and nuclear magnetic resonance (NMR) can be combined with stable isotope tracers to measure metabolic fluxes in vivo very accurately, provided appropriate analytical expertise is applied. The tracers presented herein reflect what we feel to be the most practical choices in terms of cost and utility. Although in certain cases substitutions are possible, care must be taken and careful consideration given to the fate of specific labeled carbon or hydrogen atoms.
The well-established safety and straightforward regulatory status of stable isotope tracers are worth mentioning. Stable isotopes have been safely administered in human research for decades. The long-standing policy of the US Food and Drug Administration is that “if a substance does not otherwise require submission of an IND (i.e., investigational new drug application), then that substance enriched with a stable isotope does not require an IND either. Thus, INDs are not required for metabolic tracer studies using stable isotope-enriched substances such as water, glucose and individual amino acids.” This policy is based on extensive biological evidence that the introduction of tracer levels of stable isotope-perturbed molecules carries no additional risks compared to their natural abundance congeners [7–9]. This is certainly well established for stable isotope-tagged glucose, glycerol, acetate, fatty acids, and amino acids which can be given safely orally or intravenously provided usual US Pharmacopeia (USP) precautions are taken [9].
Deuterated or heavy water is an increasingly common and highly versatile stable isotope-tagged precursor used in clinical and preclinical research [10, 11]. Deuterated water (2H2O) as a tracer for clinical studies is safe and well tolerated, as well as being simple to administer [7–9]. Deuterated water does have some biological effects at very high doses (>20 % enrichment in cellular water in an organism), but these levels are more than an order of magnitude greater than is required or achieved for flux rate measurements in people and would never be achieved in a clinical setting [12]; in general the body water enrichments (% 2H2O) used range from 1 to 2 % in clinical studies and <10 % in animal studies. The only notable side effect of giving deuterated water to humans is a transient feeling of dizziness or vertigo if the tracer is initially administered too quickly. This is thought to be the result of slight changes in the fluid dynamics in the inner ear. This effect has been reported to occur in approximately 1 in 30 subjects but can be essentially completely eliminated if the dose of deuterated water for an adult is less than 40 mL every 3 h [12].
It is recognized in the field of enzymology that there are “isotope” effects on certain reactions, comparing the rate of a 100 % labeled site vs. natural abundance. In principle this could have a small but measurable effect on stable isotope tracer studies in vivo. These theoretical isotope effects are relevant to any reaction wherein a chemical bond containing the heavy atom is broken. In vivo, however, the observed isotope effect for 13C or deuterium is minimal for almost all reactions, with the exception of certain carboxylation/decarboxylation reactions. This is confirmed by the observation that over the lifetime of a mammal, there is not a significant accumulation or depletion of heavy atom-containing molecules (as is seen in some plant metabolic processes, such as carbon dioxide fixation which may exhibit isotope effect and differential retention of 13C). Moreover, studies comparing deuterated water labeling with 13C labeling of the same pathways have repeatedly given the same results when directly compared [13] in spite of having significantly different theoretical isotope effects.
Stable isotope administration combined with mass spectrometry can reliably determine the flux rates through many metabolic pathways involved in cardio-metabolic diseases. Quantitative assessments of pathways of glucose metabolism [14, 15], fatty acid [16, 17], triglyceride [18, 19], and lipoprotein metabolism [20, 21] as well as muscle protein turnover [22, 23] including mitochondria biogenesis [24] can be studied using minimally invasive techniques applicable in early clinical as well as in preclinical studies.
De Novo Lipogenesis
Introduction
De novo lipogenesis (DNL) is the synthesis of fatty acids from their metabolic precursor, acetyl-CoA. This process can in principle occur in all cells but in humans is primarily performed, in quantitative terms, by adipose and liver tissues. Other tissues, however, such as skeletal muscle, pancreatic β-cells, skin, blood cells, and hypothalamus, have been shown to carry out DNL to some extent, with what may be critical regulatory functions in cellular metabolism. Indeed, where it was once believed to simply be an “overflow” pathway for excess carbohydrate intake, DNL is increasingly recognized to be a key regulatory pathway influencing lipid metabolism, fuel oxidation, plasma triglyceride production, tissue insulin resistance, and obesity [25, 26]. Abnormal regulation of DNL has been observed in numerous animal models of metabolic syndrome and diabetes, and recent clinical research has demonstrated increased hepatic DNL in nonalcoholic fatty liver disease (NAFLD) [27] and impaired adipose DNL in insulin resistance [28]. Finally, DNL may also play a significant role in the detrimental effects of certain dietary factors (e.g., fructose) which contribute to the etiology of cardio-metabolic diseases [29, 30]. Measurement of DNL and other lipid synthesis pathways may also be useful in the study of adipose metabolism and obesity. During the development of obesity, there is accumulation of lipid in adipose tissues. Accordingly, quantifying adipose tissue triglyceride and fatty acid synthesis is likely to be important in studying the development of obesity. The dynamic state of adipose lipid stores may play a role in the development and treatment of obesity [18].
DNL may be a viable pathway to target directly with pharmacological agents. Inhibition of DNL may increase fatty acid oxidation, relieve ectopic fat accumulation, and improve insulin sensitivity [25, 31]. Furthermore, the observation that DNL is increased in insulin resistance and NAFLD may make measurement of DNL useful for evaluating improved metabolic status in response to interventions which do not directly target lipid synthesis. It should be pointed out that simply measuring the concentrations of palmitate (the primary end product of DNL) is generally insufficient to evaluate DNL pathway activity, because DNL generally contributes a minority of circulating or stored palmitate, the majority of which is derived from diet.
There are numerous ways to approach measuring these pathways in vivo [18, 32, 33]. Here we will focus on describing a few examples of useful clinical stable isotope tracer approaches for evaluating hepatic and adipose lipid synthesis with an emphasis on the physiological and pharmacological questions they address. Specifically, two approaches to measuring lipid fluxes in vivo will be described; deuterated water labeling is useful for long-term and comprehensive analysis, and 13C-acetate labeling is an attractive option when short-term quantitative sensitivity and minimum variability of the pathway is desired.
Background
The use of stable isotope tracers to measure lipid synthesis began in the Department of Biological Chemistry of the Columbia College of Physicians and Surgeons in New York in 1935. Rudolf Schoenheimer developed the first approaches using deuterated water to measure the “dynamic state of body constituents” [34]. Many talented researchers have since utilized stable and radioactive isotopes to provide our current body of knowledge on the regulation of lipid synthesis. Contemporary measurement of DNL is performed using either deuterated water or 13C-acetate. Both of these labeling approaches have intrinsic advantages and limitations, which are highlighted below. In general, however, when utilized properly, the two methods deliver nearly identical results. While it is beyond the scope of this text to detail the analytical methods or derive calculation parameters, several excellent reviews and texts are available [3, 19, 32, 33].
Key Methods
De Novo Lipogenesis Measured Using Deuterated Water
Deuterated water can be used to assess the contribution of DNL to total palmitate in adipose samples and plasma very low-density lipoprotein (VLDL) triglycerides. Orally administered deuterated water is freely permeable across almost all biological membranes, is well mixed in the body, and its enrichment within the body water compartment is easily maintained for extended periods of time because of relatively slow turnover of body water (half-life 7–10 days). Labeled hydrogen atoms from deuterated water (2H2O) enter into biosynthetic pathways by exchange with cellular water or by specific incorporation of H from NAD(P)H. Deuterated water is a particularly useful tracer for assessment of lipid synthesis because it can be administered for long periods of time, and the label is incorporated into multiple lipid components so that, if desired, the synthesis rate of cholesterol, phospholipid, triglyceride, and fatty acids can be determined in a single experiment. Cell proliferation, i.e., deoxyribonucleic (DNA) synthesis [18, 35] as well as protein synthesis, can also be measured through deuterated water labeling [11, 22, 36] (see protein synthesis).
Deuterated water has been given to humans for more than 70 years without significant adverse effects, and animals have been raised for sequential generations on relatively high levels of deuterium water (e.g., up to 15–20 %), without phenotypic consequences or adverse effects. Additionally, no known risks have been observed in studies examining the effect of deuterated water on male or female reproductive tissues, including sperm function. As mentioned, the only minor adverse effect reported for deuterated water at these doses is transient dizziness when the initial rate of deuterated water intake is too rapid, occurring in approximately 1 in 30 individuals, which typically resolves completely within 3 h. Accordingly, care must be taken in the initial loading rate of deuterated water that is administered.
Method
Deuterated water labeling studies generally consist of several short outpatient visits for blood or tissue sampling, while the deuterated water is consumed daily at home. For proof of concept studies in healthy subjects, adult female and male volunteers are appropriate. Bulk deuterated water is available from several vendors and can be administered as 100 % or a more dilute solution, typically 70 %. Deuterated water is most conveniently prepared in single-use bottles which should be tested for sterility to ensure safe use and durable shelf life. Plasma, urine, or saliva samples are appropriate for measurement of body water enrichments provided they are collected in tubes with no additional liquid (e.g., spray-dried anticoagulants should be used). Often frequent 2H2O measurements are desired, and this can be easily done by the subject collecting saliva at home in salivette containers that are mailed to the laboratory or frozen and brought to the site during a scheduled visit. The dose of deuterated water given is dependent on the specific study design; however, a typical labeling paradigm that works well in most settings is as follows: Subjects receive loading doses of 70 % deuterated water for 2 days administered as oral doses of 50–60 mL four times daily to achieve approximately 1 % body water enrichment. Subjects then continue to take a single daily oral deuterated water dose (50–60 mL of 70 % deuterated water) until completion of the study. Subjects return to the site outpatient visits for blood collection. At all outpatient visits, plasma, urine, or saliva samples are collected to determine body deuterated water enrichments and to determine fractional DNL in very low-density lipoprotein triglyceride or total plasma triglyceride. Adipose samples can be collected after 7 days for assessment of DNL. Subcutaneous adipose tissue biopsy samples are obtained under sterile conditions and local anesthesia with 1 % lidocaine. After a small incision, a biopsy needle (Bard Biopsy Systems, Crawley, UK) is used to obtain tissue samples. The portion of adipose tissue used for lipid synthesis should be rinsed in saline and immediately frozen. Alternatively subcutaneous adipose tissue can be collected though aspiration. Examples of typical values for body water enrichment, DNL, triglyceride, and cholesterol synthesis in the plasma compartment are shown in Fig. 3.1.
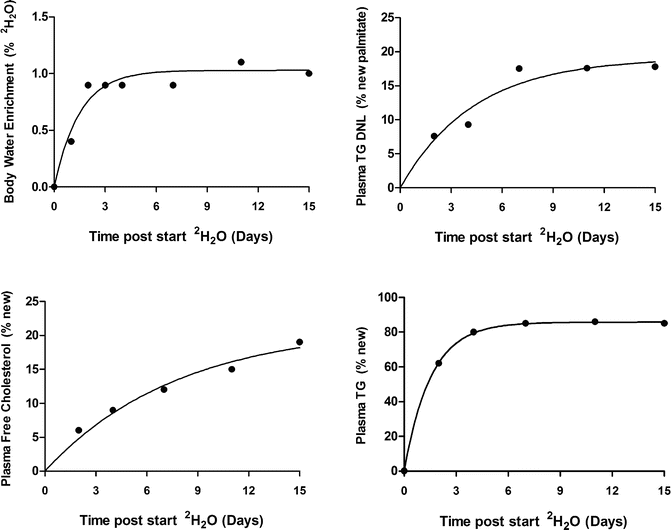
Fig. 3.1
An example of the use of oral deuterated water (2H2O) as a metabolic tracer for the simultaneous measurement of de novo lipogenesis (DNL), triglyceride (TG) and cholesterol turnover in the plasma compartment in a human volunteer. The subject received loading doses of 70 % 2H2O for 2 days (four aliquots of 60 ml of 70 % 2H2O given at least 3 h apart on day 1 and day 2) followed by a single dose of 60 ml of 70 % 2H2O once daily until the end of the experiment to achieve steady-state body deuterated water enrichments of approximately 1 %
Advantages/Disadvantages
Using deuterated water labeling for assessing de novo lipid synthesis has several intrinsic advantages over other approaches. The flexibility in timing from hours to weeks allows for measurements in different metabolic compartments to be made in the same subject. The ubiquitous nature of deuterated water labeling also allows the turnover of multiple metabolites to be assessed simultaneously. The ability to assess lipogenesis at metabolic steady state integrates diurnal and dietary fluctuations into a single real world measurement. Finally, the simple route of administration (oral, once daily) unburdens the patient and research sites from requiring overnight intravenous infusions.
There are some drawbacks to this labeling approach, principally around the interaction between behavior and metabolism. Because deuterated water labeling is generally used in the outpatient setting, potential variability or changes in diet or other behaviors need to be considered. Deuterated water labeling also requires some time to reach the optimal enrichment in the body water pool and thus is not ideal for very short-term kinetic studies that require high precursor pool enrichments. Deuterated water also has a long half-life in the body which prevents short-term (<3 weeks) restudy of subjects. For studies where very tight metabolic control over the short-term is desired, we recommend using 13C-acetate infusion method.
De Novo Lipogenesis Measured Using 13C-Acetate
Intravenous infusion of 13C-acetate is another common method used to measure hepatic DNL. Oral or intravenous acetate is efficiently taken up by the liver and the intrahepatic acetyl-CoA pool is labeled. As lipids are synthesized, 13C acetyl-CoA is incorporated into the new fatty acids and the fractional synthesis can in principle be calculated from a simple precursor/product ratio. The challenge historically has been the difficulty in accurately determining the isotope enrichment in the intrahepatic acetyl-CoA precursor pool. Additionally, other concerns regarding the lack of equilibrium of acetate across the liver have also been raised. Both of these methodological considerations have been largely resolved. The intracellular acetyl-CoA pool enrichment can be noninvasively determined using mass isotopomer distribution analysis (MIDA) [33, 37] or other similar combinatorial algorithms [38] which largely correct for any transhepatic disequilibrium [39] and have been shown to give results in line with deuterated water labeling methods where equilibration is not an issue. There are many possible ways to utilize this approach for assessing hepatic DNL. 13C-acetate can be safely infused for several days, and diurnal changes in DNL can be monitored [40, 41]. For proof of concept studies focused on the regulation of hepatic DNL, we have found the use of a controlled dietary stimulation to deliver very reproducible results in cross-sectional and particularly longitudinal studies where subjects can serve as their own controls.
While obese and diabetic subjects are known to have higher than normal basal fasting levels of hepatic fractional DNL, on the order of 15–20 % contribution to VLDL palmitate, healthy lean individuals have fasting hepatic fractional DNL on the order of 3–5 % after a brief 8–12-h infusion of labeled acetate [40, 42]. Fasting DNL varies considerably within as well as among subjects, due to influences from changes in exercise, alcohol, diet (both caloric content and carbohydrate composition) [30, 43], energy balance, weight, etc. [26]. A high carbohydrate diet can increase DNL from 5 % or less to 20 % in the fasting state, for example. Accordingly, study cohorts should be carefully enrolled to match for these physiological factors, as well as body mass index (BMI), sex, age, etc. when fasting DNL is the measurement of interest. It is known that acute ingestion of ethanol and fructose can increase fractional DNL contribution to circulating fatty acids in VLDL triglyceride from baseline to approximately 35 %, and we have shown that fructose-stimulated DNL assessments are very reproducible compared to fasting DNL [44]. The improvement in variability in the DNL measurement with fructose feeding will require fewer subjects to find certain effects in clinical studies.
Method
Subjects receive a standardized eucaloric dinner on day 0 and abstain from all foods and drinks (except water), until the oral fructose intake begins the next morning. On day 0 (e.g., 10:00 PM) a continuous infusion of 1-13C-acetic acid (sodium salt) is started and continuous until the last sample for DNL is drawn. An infusion rate of approximately 10 mg 13C-acetate per minute is ideal. A fasting blood draw is taken in the morning of day 1 prior to fructose dosing, then oral fructose (approximately 10 mg/kg fat free mass/min) is given as a drink every 30 min (e.g., 20 drinks given over 9.5 h). Blood samples are obtained regularly until 10 h post. There should be a minimum of a 5-day washout interval between fructose/[1-13C]acetate administration studies to allow labeled fatty acids to be cleared from hepatic lipid stores.
Fasting- and fructose-stimulated hepatic DNL is determined by measuring the incorporation of [1-13C] acetate into palmitate in circulating VLDL triglyceride by use of mass isotopomer distribution analysis (MIDA) [33]. VLDL is isolated from frozen plasma samples (1 mL) by ultracentrifugation. Lipoprotein triglyceride should be prepared by thin-layer chromatography (TLC), and VLDL-triglyceride fatty acids are then trans-esterified to fatty acid methyl esters in preparation for GC/MS analysis. Fractional DNL should be calculated using MIDA as described. This method for determining hepatic precursor pool enrichment is important in this setting as the acetyl-CoA pool is significantly diluted by fructose in humans. An example of changes in plasma DNL and hepatic acetyl-CoA pool dilution with fructose feeding are shown in Fig. 3.2. In practice, fructose can be replaced with other dietary components. In our hands, fructose stimulation offers a very direct and predictable model for assessing hepatic lipogenesis [44]. Mixed meal stimulation have also been shown to work well.
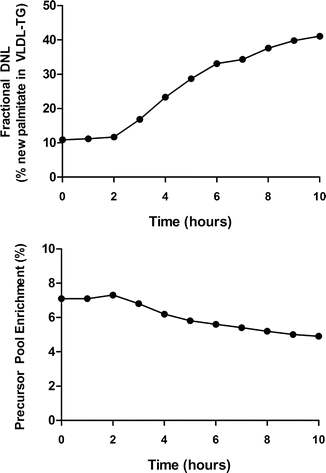
Fig. 3.2
Example of the response of hepatic de novo lipogenesis (DNL) and its precursor pool (hepatic acetyl-CoA) to acute fructose feeding in a healthy subject. The volunteer was confined to the Clinical Research Unit throughout the study under standardized conditions. At approximately 10:00 PM on day 0, an intravenous administration of [1-13C] acetate (9–9.5 mg/min) was started and continued for approximately 19.5 h. Immediately following the fasting blood draw (8:00 AM on day 1 or 0 h), oral fructose (0.25 g per kg body weight) was started (8:30 AM on day 1 or 0.5 h) and given as a drink every 30 min (20 drinks given over 9.5 h). Blood samples were obtained hourly for 10 h post fructose for the assessment of hepatic DNL
Advantages/Disadvantages
Using 13C-acetate combined with fructose administration to assess de novo lipid synthesis has several intrinsic advantages over other approaches. Because the fructose administration results in a reproducible individual response over time [44], the metabolic response to a treatment intervention such as a putative inhibitor of DNL (e.g., an acetyl-CoA carboxylase inhibitor or ATP-citrate lyase inhibitor) can be accurately quantified relative to dose and time of exposure in a small number of patients. This approach can be used for single and multiple dose studies as well as long-term interventions.
Drawbacks to this approach are largely operational; it requires overnight intravenous administration of acetate and repeated dosing of fructose during the day which can occasionally cause gastrointestinal discomfort.
Translating Results into Clinical Practice
Direct measurement of DNL can be extremely valuable in making early decisions in cardio-metabolic drug development. Just as DNL sensitively responds to diet, it can be a very sensitive barometer of metabolic changes induced by a therapeutic intervention, making it an excellent pharmacodynamic marker and in some circumstances may predict treatment response or identify candidate populations for particular therapeutic targets. The role of DNL in the pathophysiology of fatty liver, obesity, and insulin resistance all point to this being an important pathway reflecting metabolic health which is also responsive to interventions. Assessment of DNL can be useful for evaluating diabetes, fatty liver, inflammation, obesity, and dyslipidemia and is particularly informative when combined with complementary assessments such as energy expenditure, food intake, triglyceride, or lipoprotein production, to name a few.
Because the role of DNL is varied and highly context-dependent, it is a challenge to tie alterations in DNL to a single clinical endpoint. Furthermore, direct inhibitors of DNL have not been studied for the clinical pharmacodynamics of DNL reduction in relation to specific disease components. On the other hand, several studies have examined the cross-sectional relationship between DNL and disease phenotype as well as examining dietary interventions which increase DNL specifically. Using the 13C-acetate approach, Stanhope et al. [30] demonstrated that overfeeding of fructose beverages for 10 weeks increased meal-stimulated DNL by 50 % and resulted in a doubling of the postprandial triglyceride AUC (area under the curve). Interestingly, no increase was seen in fasting triglyceride or DNL in this study. Comparing insulin-resistant elderly subjects to young controls using deuterated water, Flannery et al. [45] observed a twofold higher rate of DNL in the insulin-resistant group, with corresponding increases in postprandial triglyceride concentrations, insulin level, and intrahepatic lipid content. Similarly in obese hypertriglyceridemic diabetic and nondiabetic subjects, a threefold increase in DNL was observed [40]. In a separate study, a 50 % reduction in DNL corresponded to 30 % drop in fasting plasma triglyceride. Finally, in fatty liver disease DNL has been consistently shown to be two to threefold elevated compared to controls [27]. Taking these results together it is evident that increased postprandial DNL is closely associated with clinically relevant aspects of cardio-metabolic disease.
Conclusions
DNL is a relatively straightforward metabolic pathway to measure in vivo using a variety of stable isotope methodologies. Postprandial or stimulated DNL appears to have the most relevance when translating results into clinical practice and can be performed with either 13C-acetate or deuterated water. The quantitative nature of these measurements make them well suited for early clinical trials and with the relative ease of administration of deuterated water for the study of DNL performing large clinical trials with quantitative flux measurements are now possible.
Glucose Metabolism
Introduction
Blood glucose concentrations are tightly controlled in healthy people through the regulation of both glucose disposal and glucose delivery by glucose, insulin, and other signals. In the fasting state, glucose circulating in the plasma is mainly produced by the liver whereas the brain is the major organ of glucose disposal. After a meal, glucose is absorbed from the intestine into the circulation, endogenous glucose production (EGP) is suppressed, and tissue glucose disposal is increased. Following a meal in a healthy person, glucose concentrations return to fasting levels within a few hours [46].
Type 2 diabetes is characterized by hyperglycaemia in both the fasting and the postprandial state. Even though type 2 diabetes is defined as a single disease, most clinicians recognize that type 2 diabetes is a complex, multifactorial disorder which may be the result of dysregulation of one or more pathways in glucose metabolism. Measurements of glucose concentrations are, of course, important but do not provide any mechanistic information of the metabolic events leading to type 2 diabetes. The metabolic flux of glucose through the different pathways can be measured using stable isotope tracer methodologies. These methods have been shown to be sensitive to detect changes in glucose pathways with disease and treatment, often before changes in glucose concentrations are detected. Because diabetes is such a multifactorial disease usually requiring polypharmacy for effective management, it is important to have the best possible understanding of the effect of new therapies on the metabolic pathways regulating glucose homeostasis.
Key Methods
Fasting Endogenous Glucose Production
Fasting EGP can be assessed using the isotope dilution technique [47]. Originally the method involved the use of radioactive tracers but the introduction of stable isotope-labeled glucose resulted in extensive research exploring the effect of disease and drug treatment on EGP in the postabsorptive state. Isotopic measurement of EGP represents glucose release from both the liver and the kidney, and although the quantitative contribution of renal glucose release (between 5 % and 28 % in healthy subjects) is small compared to that of the liver, one should not equate whole-body isotopically measured EGP with hepatic glucose production alone [48].
A variety of experimental protocols for the measurement of fasting endogenous glucose production have been described in the literature. Methodological differences are noted in the type of glucose tracer, administration of the tracer (continuous infusion vs. bolus), priming technique, tracer infusion time, timing, and number of sample collection and calculation models (steady-state vs. non-steady-state equations) used. It is generally accepted that the use of [U-13C6]-glucose and [6,6-2H2]-glucose provide the most relevant and easily interpretable measurement of total EGP in context of isotope loss or recycling during partial metabolism of glucose taken up by the liver [49]. Detailed description of the different tracers for the use of glucose production has been published by Wolfe [3]. Often, the tracer is administered as a primed-continuous infusion for 2–5 h, and plasma glucose enrichments are determined by GC/MS during the last 30–60 min of the steady-state period. The rate of EGP is then calculated using the steady-state equation:
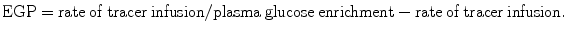 In healthy subjects a fixed priming dose corresponding to a 100-min infusion is usually used. However, in insulin-resistant states, where the glucose pool size is increased, the priming dose should be adjusted for plasma glucose concentrations, and a prolonged infusion (4–6 h) is required for valid measurements when using steady-state equations, because of slower turnover rate of the larger extracellular glucose pool. An insufficient priming and infusion time when hyperglycaemia is present has been shown to overestimate EGP due to failure to reach true plateau glucose enrichments in the plasma compartment [50–52]. Indeed, the greater EGP rates in people with type 2 diabetes compared to healthy subjects have been questioned due to tracer protocol design. In our experience, using an adjusted prime and a 5–7-h continuous infusion of [U-13C6]-glucose or [6,6-2H2]-glucose, we can reproducibly demonstrate an isotopic steady state in essentially all subjects, and we consistently observe higher EGP values in type 2 diabetes compared to healthy subjects confirming published results.
In healthy subjects a fixed priming dose corresponding to a 100-min infusion is usually used. However, in insulin-resistant states, where the glucose pool size is increased, the priming dose should be adjusted for plasma glucose concentrations, and a prolonged infusion (4–6 h) is required for valid measurements when using steady-state equations, because of slower turnover rate of the larger extracellular glucose pool. An insufficient priming and infusion time when hyperglycaemia is present has been shown to overestimate EGP due to failure to reach true plateau glucose enrichments in the plasma compartment [50–52]. Indeed, the greater EGP rates in people with type 2 diabetes compared to healthy subjects have been questioned due to tracer protocol design. In our experience, using an adjusted prime and a 5–7-h continuous infusion of [U-13C6]-glucose or [6,6-2H2]-glucose, we can reproducibly demonstrate an isotopic steady state in essentially all subjects, and we consistently observe higher EGP values in type 2 diabetes compared to healthy subjects confirming published results.
While the primed-continuous infusion of a glucose tracer is the most commonly used method for the measurement of fasting EGP, and considered the gold standard, there may be some instances where a primed constant infusion is not desirable or possible. As an alternative, a bolus injection method has been validated against the primed-continuous infusion method [53]. By this method, a glucose tracer is administered as a bolus (7 mg/ kg body weight of [U-13C6]-glucose given over 1–3 min) and frequent blood samples are collected for 1–3 h. The decay of plasma glucose tracer enrichments is fit to a two- or three-exponential curve using a kinetic simulation module (e.g., SAAM II software [54]), and EGP is then calculated from the fit parameters. While published data are currently limited to healthy volunteers, we have seen very good agreement in patients with type 2 diabetes and impaired glucose tolerance as well (unpublished results).
Several endpoints can be calculated from the direct measurement of EGP. Hepatic insulin sensitivity has been calculated as the inverse of the product of EGP and fasting plasma insulin [55], while the metabolic clearance rate (MCR), which is a measurement of the ability of whole-body tissues to take up glucose in the fasting state, is calculated as EGP divided by glucose concentration.
Sources of Fasting EGP: Gluconeogenesis and Glycogenolysis
Several approaches have been described to measure the contribution of gluconeogenesis and glycogenolysis to fasting endogenous glucose production [3]. In this chapter we focus on the method described by Hellerstein et al. involving the infusion of [2-13C1] glycerol and the use of MIDA [56, 57]. This method allows the calculation of gluconeogenesis from all three-carbon sources; lactate, amino acids, and glycerol. A primed-continuous infusion of [2-13C1] glycerol is administered to label the precursor pool (hepatic triose phosphate) for glucose synthesis for several hours (see above), and samples are taken during the last 30–60 min. The [2-13C1] glycerol can be infused with [U-13C6]-glucose or [6,6-2H2]-glucose for the simultaneous measurement of gluconeogenesis and endogenous glucose production. The principle of MIDA and its application for measuring biosynthesis of a variety of polymers (e.g., glucose) have been discussed in detail elsewhere [37, 58]. In brief, the technique consists of quantifying fractional abundances of different mass isotopomers (i.e., different isotopic isomers) in an intact polymer (e.g., glucose) that contains two or more repeats of a precursor monomeric subunit. Gluconeogenesis can be conceptualized as the polymerization synthesis of two triose-phosphate monomers. The fractional abundances of mass isotopomers (M0 for unlabeled glucose, M1 for singly labeled glucose, and M2 for doubly labeled glucose) in the polymer must then be accurately measured by GC/MS and compared with the natural fractional abundance of baseline values to calculate changes, or excesses, of mass isotopomeric species in the polymer. The precursor pool enrichment (i.e., the hepatic triose-phosphate isotope content, for glucose) can then be back-calculated from the pattern or distribution of mass isotopomeric excesses in the polymer (glucose), using formulae derived from the binomial or multinomial expansions, as described in detail elsewhere [57]. Once the precursor pool enrichment is established, the precursor/product relationship can be applied for calculation of the fraction of polymers that were derived from the endogenous biosynthetic pathway during the labeling period. In the case of gluconeogenesis, the ratio of excess doubly labeled glucose to excess single-labeled glucose species reveals the isotopic abundance of the true precursor pool, from which fractional gluconeogenesis can be calculated. Absolute flux through gluconeogenesis into plasma glucose can then be calculated as the fractional gluconeogenic contribution times the rate of fasting EGP. These two parameters can be assessed simultaneously, as described above. The absolute contribution of glycogenolysis to fasting EGP is calculated as the difference of rate of fasting EGP and rate of gluconeogenesis. Potential methodological problems have been addressed and discussed in detail by Neese et al. [57]. More complex stable isotope techniques like the secreted glucuronate technique have been described for the measurement of intrahepatic glycogen fluxes. This method has been described in detail elsewhere [57, 59].
Suppression of EGP During a Hypersulinaemic Euglycaemic Clamp
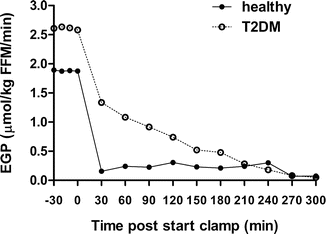
Hepatic insulin resistance is a key player of whole-body insulin resistance in people with type 2 diabetes [60]. Fasting rates of EGP are often elevated and suppression of glucose production during intestinal absorption of a meal is impaired in type 2 diabetes. The gold standard to determine hepatic insulin sensitivity is the measurement of partial suppression of EGP during a low-dose insulin infusion. Tracer methodology combined with the hyperinsulinaemic euglycaemic clamp allows the simultaneous measurement of fasting EGP, insulin-mediated suppression of hepatic glucose production (hepatic insulin sensitivity), and tissue glucose disposal (peripheral insulin sensitivity) independent of the effects of hyperglycaemia per se. Currently the best methodology to do this is the so-called hot GINF (glucose infusion) method proposed a few decades ago [61, 62]. The method is designed to keep the ratio of the glucose tracer to tracee in plasma constant during the fasting state and during the clamp to minimize errors (or negative values) in EGP measurements due to non-steady-state conditions. Ideally, deviations in circulating tracer enrichments should be kept to <20 % to avoid errors in the EGP results [63]. The method involves the administration of a glucose tracer as a primed-continuous infusion and as part of the exogenous glucose infusion (GINF) used during the clamp. The tracer enrichment of the GINF is calculated to be equal to the expected plasma tracer enrichment at the start of clamp. Originally a radioactive or hot glucose tracer was added to the exogenous glucose infusion, hence the name hot GINF. The continuous infusion of the glucose tracer is started before the clamp to determine fasting EGP as described above. Its rate is then reduced at the start of the clamp and after fasting EGP measures are completed to approximate the expected fall of EGP during the clamp (e.g., the infusion rate of the tracer is reduced to half the starting rate if a 50 % reduction of EGP is expected during the clamp). The anticipated fall in EGP during the clamp depends on several factors such as patient population (e.g., healthy subjects may have a greater suppression of EGP than subjects with type 2 diabetes under similar insulin conditions), clamp insulin infusion rates, and duration of the clamp. Pilot experiments are recommended to determine the optimal tracer design during the clamp if there is not prior experience with the patient population and method. An intrinsic limitation of this approach is therefore that the quantitative effects of an intervention on EGP have to be to some extent known in advance to apply the model. Examples of EGP measured during a clamp in a healthy subject and an individual with type 2 diabetes are shown in Fig. 3.3.
 Regulatory Considerations for Early Clinical Development of Drugs for Diabetes, Obesity, and Cardiometabolic Disorders
Regulatory Considerations for Early Clinical Development of Drugs for Diabetes, Obesity, and Cardiometabolic Disorders
 Early Phase Metabolic Research with Reference to Special Populations
Early Phase Metabolic Research with Reference to Special Populations
 Omics: Potential Role in Early-Phase Drug Development
Omics: Potential Role in Early-Phase Drug Development
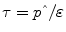 Computational Modelling of Energy Metabolism and Body Composition Dynamics
Computational Modelling of Energy Metabolism and Body Composition Dynamics
 Measurement of Energy Expenditure
Measurement of Energy Expenditure
 Assessment of Body Composition
Assessment of Body Composition




Related posts:
Regulatory Considerations for Early Clinical Development of Drugs for Diabetes, Obesity, and Cardiometabolic Disorders
Early Phase Metabolic Research with Reference to Special Populations
Omics: Potential Role in Early-Phase Drug Development
Measurement of Energy Expenditure
Assessment of Body Composition
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
