Iron deficiency anemia is a common global problem whose etiology is typically attributed to acquired inadequate dietary intake and/or chronic blood loss. However, in several kindreds multiple family members are affected with iron deficiency anemia that is unresponsive to oral iron supplementation and only partially responsive to parenteral iron therapy. The discovery that many of these cases harbor mutations in the TMPRSS6 gene led to the recognition that they represent a single clinical entity: iron-refractory iron deficiency anemia (IRIDA). This article reviews clinical features of IRIDA, recent genetic studies, and insights this disorder provides into the regulation of systemic iron homeostasis.
Key points
- •
Iron-refractory iron deficiency anemia (IRIDA) is an inherited disorder of systemic iron balance in which both absorption and utilization of iron are impaired.
- •
Patients with IRIDA show iron deficiency anemia that is refractory to oral iron therapy but partially responsive to parenteral iron.
- •
IRIDA is caused by mutations in the gene TMPRSS6 .
- •
TMPRSS6 encodes matriptase-2, a transmembrane serine protease expressed by the liver that regulates the production of the iron regulatory hormone hepcidin.
- •
Studies conducted in tissue culture systems and mouse models have enhanced our understanding of the underlying pathogenesis.
Introduction
Iron is an essential metal for many biologic processes in mammals. Its primary role is to bind oxygen in the heme moiety of hemoglobin. Iron also plays a central role in the enzymatic transfer of electrons performed by cytochromes, peroxidases, ribonucleotide reductases, and catalases. This reactivity of iron also has the potential to cause damage to biologic systems if iron is “free” and not bound and transported by a finely regulated and complex system of proteins that maintain iron homeostasis.
Under normal physiologic conditions in the adult male, only 1 to 2 mg of the 20 to 25 mg of iron required daily to maintain erythropoiesis enters the body through carefully regulated intestinal absorption. Most of the daily iron need is derived from the recycling of erythroid iron through phagocytosis of senescent red cells by reticuloendothelial macrophages and degradation of hemoglobin. As humans have no physiologically regulated mechanism for excreting iron from the body, control of iron balance occurs almost entirely at the level of intestinal absorption.
Virtually all plasma iron exists bound to the circulating glycoprotein transferrin (TF), which allows the iron to remain soluble, renders iron nonreactive, and facilitates its cellular import through the transferrin cycle. Iron-loaded plasma transferrin binds to transferrin receptors (TFR) on the cell surface. The TF/TFR receptor complex is endocytosed, and acidification of the endosome results in the release of iron from TF. The iron is transported out of the endosome into the cytoplasm, and the empty TF and TFR return to the cell surface and are released into the plasma to repeat this cycle.
Most nonerythroid intracellular iron is stored in hepatocytes and macrophages in the form of ferritin, a multimeric iron storage protein whose structure facilitates iron bioavailability in response to cellular need. Intracellular iron, either absorbed by the duodenal enterocyte or liberated by macrophages from heme recycling, is either stored as ferritin or exported into the plasma by ferroportin. Ferroportin, which is the sole known mammalian cellular iron exporter, is highly expressed on the basolateral membrane of enterocytes and on the cell membrane of reticuloendothelial macrophages.
Iron homeostasis requires carefully coordinated regulation of intestinal iron absorption, cellular iron import/export, and iron storage. Hepcidin, a small circulating peptide released by the liver, is the master regulator of systemic iron balance. Hepcidin limits both iron absorption from the intestine and iron release from macrophage stores by binding to ferroportin and triggering ferroportin’s internalization and degradation. Hepcidin expression is modulated in response to several physiologic and pathophysiological stimuli, which include systemic iron loading, erythropoietic activity, and inflammation.
As is the case with many physiologic processes, spontaneous mutations leading to disease in animals and humans have revealed much about the normal regulation of iron transport and storage in humans. In particular, the identification of TMPRSS6 as the gene mutated in cases of iron-refractory iron deficiency anemia (IRIDA), has increased our knowledge of the molecular mechanisms that regulate hepcidin expression.
Introduction
Iron is an essential metal for many biologic processes in mammals. Its primary role is to bind oxygen in the heme moiety of hemoglobin. Iron also plays a central role in the enzymatic transfer of electrons performed by cytochromes, peroxidases, ribonucleotide reductases, and catalases. This reactivity of iron also has the potential to cause damage to biologic systems if iron is “free” and not bound and transported by a finely regulated and complex system of proteins that maintain iron homeostasis.
Under normal physiologic conditions in the adult male, only 1 to 2 mg of the 20 to 25 mg of iron required daily to maintain erythropoiesis enters the body through carefully regulated intestinal absorption. Most of the daily iron need is derived from the recycling of erythroid iron through phagocytosis of senescent red cells by reticuloendothelial macrophages and degradation of hemoglobin. As humans have no physiologically regulated mechanism for excreting iron from the body, control of iron balance occurs almost entirely at the level of intestinal absorption.
Virtually all plasma iron exists bound to the circulating glycoprotein transferrin (TF), which allows the iron to remain soluble, renders iron nonreactive, and facilitates its cellular import through the transferrin cycle. Iron-loaded plasma transferrin binds to transferrin receptors (TFR) on the cell surface. The TF/TFR receptor complex is endocytosed, and acidification of the endosome results in the release of iron from TF. The iron is transported out of the endosome into the cytoplasm, and the empty TF and TFR return to the cell surface and are released into the plasma to repeat this cycle.
Most nonerythroid intracellular iron is stored in hepatocytes and macrophages in the form of ferritin, a multimeric iron storage protein whose structure facilitates iron bioavailability in response to cellular need. Intracellular iron, either absorbed by the duodenal enterocyte or liberated by macrophages from heme recycling, is either stored as ferritin or exported into the plasma by ferroportin. Ferroportin, which is the sole known mammalian cellular iron exporter, is highly expressed on the basolateral membrane of enterocytes and on the cell membrane of reticuloendothelial macrophages.
Iron homeostasis requires carefully coordinated regulation of intestinal iron absorption, cellular iron import/export, and iron storage. Hepcidin, a small circulating peptide released by the liver, is the master regulator of systemic iron balance. Hepcidin limits both iron absorption from the intestine and iron release from macrophage stores by binding to ferroportin and triggering ferroportin’s internalization and degradation. Hepcidin expression is modulated in response to several physiologic and pathophysiological stimuli, which include systemic iron loading, erythropoietic activity, and inflammation.
As is the case with many physiologic processes, spontaneous mutations leading to disease in animals and humans have revealed much about the normal regulation of iron transport and storage in humans. In particular, the identification of TMPRSS6 as the gene mutated in cases of iron-refractory iron deficiency anemia (IRIDA), has increased our knowledge of the molecular mechanisms that regulate hepcidin expression.
Clinical presentation
In 1981, Buchanan and Sheehan described 3 siblings with iron deficiency anemia despite adequate dietary iron intake and no evidence of gastrointestinal blood loss. All 3 failed to respond to oral ferrous sulfate therapy. In 2 of the siblings, a formal oral iron “challenge” (see Box 2 ) to assess for impaired intestinal iron absorption failed to show evidence of a rise in serum iron 2 hours after the oral administration of 2 mg/kg elemental iron as ferrous sulfate. Following intramuscular injection of iron dextran, the 3 siblings also showed only a partial hematological response assessed by hemoglobin and red cell indices. In addition, although intramuscular iron administration raised the serum ferritin level, suggesting restoration of iron stores, the patients nevertheless remained hypoferremic. The investigators postulated that the phenotype was explained in part by an inherited, iron-specific absorptive defect, which was further compounded by a defect of iron utilization reflected in the partial response to parenteral iron therapy.
Pearson and Lukens subsequently described 2 affected siblings. In addition to recognizing the intestinal iron uptake defect reflected in the failed response to oral iron challenge, these investigators also documented a discordance in the rate of decline of transferrin saturation (rapid) and serum ferritin (slower) after iron dextran administration. Given that iron dextran must be phagocytosed and “recycled” by macrophages before the iron can be made available for erythropoiesis, the investigators postulated that a macrophage iron retention phenotype/macrophage iron recycling defect contributed to the phenotype.
Further cases of familial iron deficiency anemia with similar clinical presentations were subsequently reported, which provided additional insight into the mode of genetic transmission as well as the underlying pathophysiological defect. Brown and colleagues reported 2 affected female siblings of Northern European ancestry whose parents exhibited normal hematological parameters, thus suggesting a recessive mode of transmission for the disorder. Galanello and colleagues provided additional evidence for autosomal recessive transmission in a large kindred that originated in a small village in southern Sardinia; the structure of this pedigree, which contained 5 affected individuals, suggested that the disorder might be caused by homozygosity for a mutation that arose in a common ancestor. Hartman and Barker reported an affected African American sibling pair in whom bone marrow biopsies performed after parenteral iron therapy failed to demonstrate normal sideroblasts (erythroid normoblasts containing stainable nonhemoglobin iron in the cytoplasm), despite the presence of stainable iron in bone marrow macrophages. This observation thus provided evidence in support of the iron utilization defect that had been postulated by Buchanan and Sheehan.
Review of these case reports identified several unifying features that suggested that these cases represented the same underlying disorder, a condition that has been termed iron-refractory iron deficiency anemia (IRIDA). These key features of IRIDA include ( Box 1 ) (1) congenital hypochromic, microcytic anemia (hemoglobin 6–9 g/dL); (2) very low mean corpuscular volume (45–65 fL); (3) very low transferrin saturation (<5%); (4) abnormal oral iron absorption (as indicated by a lack of hematological improvement following treatment with oral iron or failure of an oral iron challenge); (5) abnormal iron utilization (as indicated by a sluggish, incomplete, and transient response to parenteral iron); and (6) an inheritance pattern compatible with autosomal recessive transmission. In these cases, acquired causes of iron deficiency (eg, gastrointestinal blood loss) and inherited causes of microcytosis (eg, thalassemias, lead toxicity) were excluded by extensive laboratory testing. Furthermore, no case showed clinical evidence of a chronic inflammatory disorder or generalized intestinal malabsorptive defect. Hemoglobinopathies and sideroblastic anemias were excluded as potential causes of the microcytosis by hemoglobin electrophoresis and bone marrow examinations, respectively.
- •
Lifelong/congenital, usually presents in childhood
- •
Severe microcytosis (mean corpuscular volume 45–65 fL)
- •
Moderate/severe anemia (hemoglobin 6–9 g/dL)
- •
Severe hypoferremia with very low transferrin saturation (<5%)
- •
No or minimal response to oral iron supplementation
- •
Abnormal oral iron absorption/failure of an oral iron challenge
- •
Incomplete and transient response to parenteral iron
- •
Autosomal recessive transmission
- •
Anemia often ameliorates into adulthood, although hypoferremia persists
In the published IRIDA cases, subjects were generally healthy and growing normally, and the anemia was typically detected during routine screening usually conducted before the age of 2 years. Thus, from the clinical histories, it was not known if patients with IRIDA were already iron-deficient at birth. Of note, the proband reported by Brown and colleagues, who was diagnosed with microcytic anemia at age 9 months, showed plentiful reticuloendothelial iron stores on an initial bone marrow examination performed after a 3-month failed course of oral iron therapy but showed an absence of stainable iron on repeat bone marrow examination performed at age 4 years after a course of intramuscular iron. These findings, along with reports of normal birth weights for IRIDA patients, raise the possibility that in utero iron transfer may be normal in these patients, with the depletion of iron stores occurring only after birth.
For unclear reasons, the clinical signs and symptoms of IRIDA appear distinct from severe acquired iron deficiency anemia. Although IRIDA subjects have laboratory evidence of severe iron deficiency, clinical signs observed in acquired iron deficiency have been noted inconsistently in the reported IRIDA cases. Moderate to severe pallor was described in the kindred reported by Melis and colleagues. The 18-month-old proband reported by Andrews was described as pale with dry skin. Studying the kindred originally reported by Brown and colleagues, Pearson and Lukens noted that the affected siblings developed angular cheilitis (crusted, painful lesions at corners of their mouths) that receded after intravenous iron therapy. Only rarely are signs and symptoms associated with iron deficiency, such as koilonychias or hair loss, described in IRIDA. Although IRIDA has been considered a rare clinical entity based on the small number of cases reported in the literature, it is possible that in the absence of routine laboratory screening for anemia, many cases never come to clinical attention because of the normal growth and development of the affected individuals. Remarkably, despite congenital, severe iron deficiency, long-term follow-up of the affected subjects has shown normal growth and normal intellectual development, with no evidence of the cognitive concerns on which iron deficiency screening in infancy have been founded.
Given the small number of reported cases, experience with the natural history and long-term management of IRIDA is, at present, limited. Pearson and Lukens proposed a treatment regimen that involved the parenteral administration of iron dextran every 2 to 4 years, or when serum ferritin levels fell below 50 to 75 ng/mL or the mouth ulcerations observed in their patients recurred. Hartman and colleagues described the course of 5 patients with IRIDA who had been followed for 15 years. They noted that repeated iron infusions that elevated the serum ferritin to levels greater than 200 ng/mL resulted in considerable improvement in both the anemia and microcytosis. Although the serum iron and transferrin saturation occasionally reached the normal range, the patients generally developed recurrent hypoferremia. With the cessation of iron infusions, microcytosis returned, but not to the severe degree present in infancy. In the affected family members studied by Galanello and colleagues (who on last report range from 18 to 48 years of age), the anemia was more severe during childhood, requiring intermittent intravenous iron administration. However, hemoglobin levels of 10.0 to 13.9 g/dL were maintained in the adult affected subjects, although laboratory findings of iron-restricted erythropoiesis (low mean corpuscular volume, low mean corpuscular hemoglobin, low serum iron, low transferrin saturation) persisted. In addition to a relative amelioration of anemia into adulthood, serum ferritin levels also appeared to rise with age in this kindred. The investigators suggested that the increased severity of the anemia during childhood could indicate the greater iron demands for body growth and for the accompanying expansion of the red cell mass that occurs during this period; in adulthood, however, a larger proportion of the limited iron available could be used in erythropoiesis.
Genetics
Strong evidence that the IRIDA phenotype has an inherited basis was obtained through genetic characterization of a large, consanguineous kindred from Sardinia. In this kindred, in which disease in affected individuals could be attributed to homozygosity for a mutation arising in a common ancestor, the IRIDA phenotype mapped to the long arm of chromosome 22 (22q12.3–13.1) under a model of recessive inheritance. IRIDA subsequently was shown to be caused by mutations in the gene TMPRSS6 , which resides within this critical region of chromosome 22q and for which a key role in iron balance had recently been revealed through study of the orthologous gene in mice (see later in this article). Notably, patients with the IRIDA phenotype showed levels of hepcidin in their serum, plasma, and/or urine that were indicative of impaired hepcidin regulation. Although hepcidin levels are normally reduced in response to systemic iron deficiency (an adaptive response to promote absorption of dietary iron), patients with IRIDA displayed hepcidin levels that were either within or above the reference range. Given the known ability of hepcidin to limit ferroportin-dependent iron export from enterocytes and macrophages, the inappropriately elevated hepcidin levels in IRIDA provide insight into the iron refractory features of the disorder. Specifically, the inappropriate hepcidin excess in IRIDA can explain (1) the development of systemic iron deficiency as a result of impaired absorption of dietary iron, (2) the failure to achieve a hematological response to oral iron therapies, and (3) the sluggish and incomplete utilization of parental iron formulations, which consist of iron-carbohydrate complexes that require processing by macrophages before the iron can be used in erythropoiesis. In many respects, IRIDA can be considered the pathophysiologic and phenotypic opposite of hereditary hemochromatosis, in which the “uncoupling” of appropriate hepcidin expression from the sensing of iron stores results in an inappropriate hepcidin “deficiency” (see the article by Brissot, elsewhere in this issue).
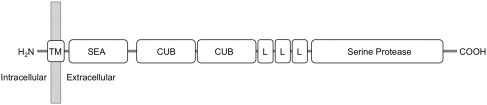
TMPRSS6 (transmembrane protease, serine 6) encodes matriptase-2, a membrane-spanning protease that is primarily expressed by the liver. Matriptase-2 is a member of the type II transmembrane serine protease (TTSP) family, a group that is anchored to the membrane at their amino termini. The protein name, matriptase-2, reflects structural homology to another TTSP, matriptase-1. Matriptase-2 contains a large extracellular region containing several structural domains, including an SEA (sea urchin sperm protein, enteropeptidase, agrin) domain, 2 CUB (C1r/C1s, urchin embryonic growth factor, bone morphogenetic protein 1) domains, 3 LDLRA (low-density lipoprotein receptor class A) domains, and a C-terminal catalytic domain containing a classic catalytic triad of serine, histidine, and aspartic acid residues ( Fig. 1 ). Matriptase-2 is believed to be synthesized as an inactive, membrane-bound, single-chain polypeptide that undergoes a complex series of proteolytic cleavage events during zymogen activation. When overexpressed in cultured cells, matriptase-2 localizes to the plasma membrane and is shed from the cell surface as an activated, 2-chain form. Recombinant matriptase-2 has been shown in vitro to degrade components of the extracellular matrix and basement membrane, such as fibrinogen, fibronectin, and type I collagen. Interestingly, overexpression of matriptase-2 in breast and prostate cancer cell lines can reduce their invasive properties in vitro, suggesting a possible role for matriptase-2 in cancer development and progression.
A key role for TMPRSS6 in iron homeostasis was first revealed through elucidation of the genetic basis of a chemically induced, recessive mutant mouse phenotype termed mask . The mask mutant received its name because it showed progressive loss of truncal hair but retained hair on the head. Notably, mice with the mask phenotype also exhibited microcytic anemia, low plasma iron levels, and low iron stores when raised on a standard rodent laboratory diet. Furthermore, mask mice showed evidence of defective hepcidin regulation in the setting of iron deficiency. Although control mice suppressed hepatic hepcidin production in response to a low-iron diet (an appropriate physiologic response to promote iron absorption), hepcidin messenger RNA (mRNA) levels in livers of mask mutants were inappropriately elevated. Genetic mapping of the underlying mutation revealed that mask mice were homozygous for a mutation that resulted in defective splicing of the Tmprss6 transcript, which eliminated the proteolytic domain of matriptase-2. The elevated hepcidin mRNA levels detected in the livers of the mask mutants suggest that the normal function of the Tmprss6 gene product, matriptase-2, is to lower hepcidin expression by the liver.
A Tmprss6 knockout ( Tmprss6 –/– ) mouse, generated by standard gene-targeting techniques, exhibited a phenotype very similar to the mask Tmprss6 splicing mutant, including the key feature of hepatic hepcidin overexpression. Notably, the anemia and alopecia phenotypes of both the engineered and the chemically induced Tmprss6 mutants could be rescued by iron administration. Consistent with the known ability of hepcidin to promote ferroportin internalization and degradation, duodenal enterocytes of Tmprss6 –/– mice showed decreased ferroportin protein expression in the basolateral membrane that was accompanied by histologic evidence of iron retention within these cells. Thus, in the setting of hepcidin elevation, when basolateral export of iron into the plasma is restricted, iron accumulates within duodenal enterocytes and is ultimately lost from the body when these cells are shed into the gut lumen.
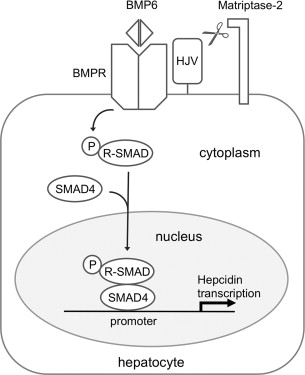
Studies conducted in tissue culture systems and transgenic models have begun to shed insight into the mechanism by which matriptase-2 regulates hepcidin production. This mechanism appears to involve modulation of bone morphogenetic protein (BMP)/SMAD signaling, a key signal transduction pathway that promotes hepcidin transcription in hepatocytes ( Fig. 2 ). BMPs are secreted ligands of the transforming growth factor β superfamily that interact with type 1 and type 2 BMP receptors at the cell membrane to trigger the phosphorylation of multiple receptor-associated SMAD proteins (SMAD1, SMAD5, SMAD8). Once phosphorylated, these receptor-associated SMADs bind to a common mediator, SMAD4, forming heterodimeric complexes that translocate to the nucleus to regulate transcription of BMP target genes, including the gene encoding hepcidin, by binding to specific elements in the promoters of genes. Of note, signaling through the BMP pathway is modulated in response to hepatic iron stores. When local iron stores increase, the liver raises expression of BMP6, the particular BMP ligand that appears to play a key role in promoting hepcidin transcription. This leads to increased hepcidin expression, an adaptive response to limit further iron absorption from the diet.