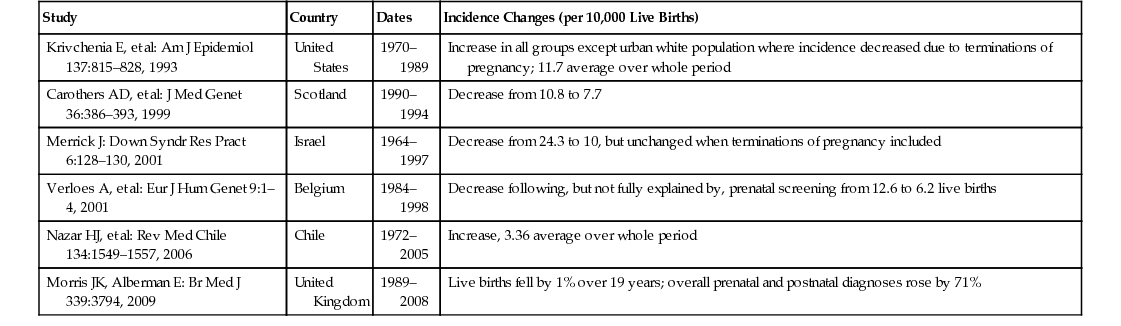
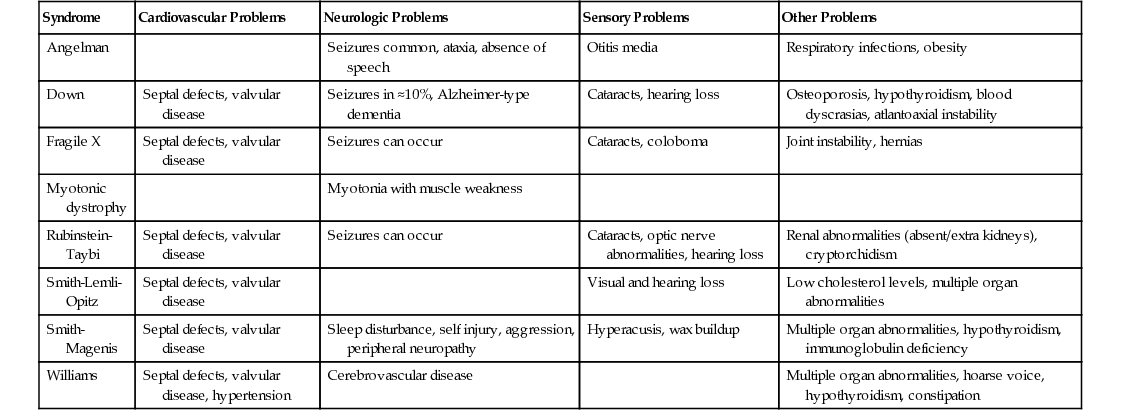
John M. Starr Intellectual disability (ID) is the current term used to describe what in the United Kingdom has been known as learning disability and in the United States as mental retardation. The World Health Organization’s International Classification of Diseases (ICD-10) still uses the term mental retardation, and its report on healthy aging in this population uses the term intellectual disabilities. In Australia the 1986 Victorian Act of Parliament defines intellectual disability in the following way: Intellectual disability in relation to a person over the age of five years means a significant sub-average general intellectual functioning existing concurrently with deficits in adaptive behavior and manifested during the developmental period. (Intellectually Disabled Persons Services Act, 1986) The threshold at which general intellectual functioning is considered “subaverage” is often fixed at an IQ of 70, two standard deviations below the mean IQ. Controversially in 1992 the American Association on Intellectual and Developmental Disabilities (AAIDD) loosened this threshold to include people with IQs in the range of 70 to 75. The AIDD also required deficits in 2 out of 10 assessed areas of adaptive functioning. This definition was adapted by the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders, fourth edition (DSM-IV). In 2002 the AAIDD reinstated the IQ 70 threshold and required deficits in conceptual, social, and practical adaptive skills to be present. Areas covered by these skills include communication, personal care, home life, social skills, community utilization, self-governance, health and safety, functional academic skills, work, and leisure activities. These changes in definition have implications for epidemiologic data collection, but the key concept of ID remains. An IQ less than 70 is necessary but is, in itself, inadequate for the diagnosis to be made. For the diagnosis to be made, there must be evidence of both a developmental disorder (with onset during childhood) and deficits in adaptive behavior. Further classification of ID can be made within the broad definition. The Diagnostic Criteria for Psychiatric Disorders for Use with Adults with Learning [Intellectual] Disabilities (DC-LD)1 describes the mental health of a person with ID in terms of ID severity, ID causes, and related mental disorders (developmental disorders, psychiatric illness, personality disorders, problem behaviors, and other disorders). Severity is grouped according to IQ: 50 to 69, mild ID; 35 to 49, moderate; 20 to 34, severe; and less than 20, profound. The Swedish model of ID classification developed by Kylen2 is often helpful in clinical situations where IQ is not known: Severity may also be broadly estimated in terms of functional abilities: Mild: Social and work skills adequate to work at a minimum wage Moderate: Requires significant support to be able to work in a protected environment Severe: Can partially contribute to his or her economic support with total supervision In addition, the DC-LD includes appendices that relate to medical factors influencing health status and contact with health services. The latter are highly relevant because developmental disorders that affect the brain, giving rise to ID, often affect other body systems also. The cause of ID is frequently unknown in older adults but can be considered along conventional lines of external causes (infection, injury, poisoning), internal disorders (endocrine, metabolic), perinatal insults, and congenital conditions (chromosomal abnormalities, gene mutations). The latter are of particular relevance to the health of older adults with ID as specific syndromes are associated with risk of particular physical disorders and diseases. Common syndromes seen in older adults include Down syndrome (DS), Angelman syndrome, fragile X syndrome, Klinefelter syndrome, Turner syndrome, and Williams syndrome. Table 57-1 provides a brief description of these. It is worth noting that, given the preceding definition of ID, by no means does everyone with one of these syndromes fulfill the diagnostic criteria for ID; this is particular true of women with Turner syndrome, who have a tendency for nonverbal cognitive deficits but are often of average intelligence. TABLE 57-1 Characteristics of Common Syndromes Associated With Intellectual Disability X-linked, semi-dominant disorder with reduced penetrance Number of fragile sites on X-chromosome identified, two important for intellectual disability around X27.3 Just as there is a considerable overlap between congenital syndromes, such as DS and ID, there is a similar overlap between ID and autism. The diagnosis of autism depends on (1) abnormal social development, (2) communication deficits, and (3) restricted and repetitive interests and behavior. Approximately three quarters of people with autism have a nonverbal IQ less than 70 and hence also fulfill diagnostic criteria for ID, but in autism social and communication skills are worse than expected for any given nonverbal IQ. In 2001 the World Health Organization reported, The prevalence figures [of ID] vary considerably because of the varying criteria and methods used in the surveys, as well as differences in the age range of the samples. The overall prevalence of mental retardation is believed to be between 1% and 3%, with the rate for moderate, severe and profound retardation being 0.3%.3 Extrapolating these figures to the United Kingdom provides estimates of approximately 175,800 people with moderate-to-profound ID and between 586,000 and 1,465,000 with mild ID.4 For Finland the equivalent estimates are 15,300 and between 51,000 and 127,500, respectively. Population-based surveys in Finland have estimated moderate-to-profound ID prevalence at no more than 0.2% and overall ID prevalence at just over 1%.4 The situation is similar for the United Kingdom.4 Notably, Finnish prevalence rates estimated from national registers is a little lower at 0.7%, perhaps indicating that not all people with ID are known to Finnish health or social services.5 Within overall prevalence figures there is considerable variation by age. In the Finnish national register survey, the rates were 0.53% for individuals aged 15 years and younger, 0.70% for those aged 16 to 39 years, 0.92% for those 40 to 64 years old, and 0.38% for those 65 years and older.5 Variation in Finland between age groups was attributed to changes in incidence, mortality, diagnostic practices, and benefit provision. Changes in diagnostic practices have been discussed in the previous section and benefit provision is specific to Finland, but changes in ID incidence and mortality have been tracked across the world. A meta-analysis of 52 population-based studies also estimated prevalence at just over 1%, falling with age.6 Estimation of ID incidence is problematic given that ID is, by definition, a developmental disorder and thus there is no single point at which it is recognized. In view of this, DS is often used as a proxy because it is the largest single cause of ID. However, its use as a proxy is far from ideal because risk is clearly associated with maternal age and consequent prenatal screening that has been widely introduced. Table 57-2 summarizes secular trends in DS incidence from various countries. Overall DS incidence appears to have been rising prior to the introduction of prenatal screening. This resulted in a decrease that is projected to be offset by increasing mean maternal age. Overall, there seems little to indicate a great change in ID incidence per 1000 live births, and numbers of people with ID may track the overall birth rates in different countries. Mortality has had the greatest impact on ID prevalence, especially in older age groups. In 1900 a child with DS would expect to survive to approximately 9 years of age. In the United States median age at death for children with DS increased from 25 years in 1983 to 49 years in 1997.7 Mean life expectancy for a child born with DS in the United Kingdom in 2011 was 51 years, with a median life expectancy of 58 years.8 The improvement likely reflects improved socioeconomic circumstances, improved correction of congenital cardiac abnormalities, and perhaps changing attitudes to treating people with ID. There is only a minor difference in life expectancy for people with DS compared with other causes of ID.9 Identification of common causes of death in people with ID is made difficult by poor death certificate completion. For example, many people in the United States had either ID or DS listed as a primary cause of death, which is inappropriate.9 However, as the ID population ages, the causes of death are thought to resemble those in the general population more and more closely. Those with mild ID survive longer, and there is some equivalence to this observation in the general population, where people with low IQs within the normal range suffer premature mortality largely attributable to cardiovascular disease.10 Current trends suggest that people with ID can expect to live to 60 to 65 years, and an increasing proportion will survive beyond this age. Even with recent improvements, the life expectancy of people with ID is considerably less than that of the non-ID population. This raises the question as to whether ID is associated with accelerated biologic aging. The measurement of biologic age usually depends on identifying suitable biomarkers of aging. Criteria for such biomarkers have been proposed11: 1. They must reflect some basic biologic process of aging rather than disease. 2. They must have high cross-species reproducibility. 3. They must change independently of chronologic time. 4. They must be obtainable antemortem. 5. They must be measurable over a short period compared to the life span of the organism. One such biomarker is telomere length.12 There is a paucity of data on telomere length in ID in general, but there is evidence for telomere shortening in DS13 though this appears to be downstream from cellular redox status14 as is also likely in the general population.15 In cri-du-chat syndrome there is often a deletion of the short arm of chromosome 5 where the telomerase reverse transcriptase (hTERT) gene is localized (5p15.33). Reconstitution of telomerase activity by ectopic expression of hTERT extends telomere length, increases population doublings, and prevents the end-to-end fusion of chromosomes.16 It may thus be one element contributing to the syndrome’s phenotypic features. Whether this is the case or not, accelerated telomere shortening occurs with aging in this syndrome.17 At least a further 5% of ID is attributable to similar subtelomeric deletions or copy number variations18 and these, too, can influence telomere length. Telomere length is thus a potentially useful index of accelerated biologic aging in ID, but whether it contributes to aging itself or is only a correlate remains unclear. Moreover, telomere shortening is subject to syndrome-specific effects. Beyond the cellular level, various physiologic biomarkers are also affected in ID. Physiologic variables are long recognized as indices of biologic age.19 The limited data available indicate that people with DS have accelerated biologic aging but that people with nonsyndromic ID do not.20,21 In summary, the evidence suggests that syndromic biologic aging dominates over any accelerated biologic aging that might be associated with ID in general and that different ID syndromes are likely to have different aging profiles depending on the specific genetic changes underlying them. People with ID exhibit considerable morbidity. A study of 346 people aged 20 to 50 years in North Sydney found they had a mean 2.5 major problems and 2.9 minor problems each with 42% of these undiagnosed prior to the study, and of the 58% already known, only 49% were being managed adequately.22 A study of 1371 adults aged 40 years and older in New York State found that increased age was associated with higher prevalence of cardiovascular disease, cancer, respiratory disease, musculoskeletal disorders, infections, and visual and hearing impairments; gastrointestinal disease was not associated with age but with being male, more severe ID, cerebral palsy, and obesity.23 Compared with the non-ID population, there was less cardiovascular disease and musculoskeletal disease, except osteoarthritis, and people with DS did not have solid neoplasias. However, these data may reflect underdiagnosis and lifestyle factors such as the low rate of cigarette smoking among people with ID. A similar age-related pattern of disease was found in southern Holland.24 The pattern may change as fewer people with ID live in institutions: there is evidence from the United Kingdom that poor diet, reduced physical activity, and obesity risk factors are greater in women with ID who are more able and independent.25 In addition to the general tendency for high levels of morbidity, which is associated with both age and degree of ID severity, specific syndromes carry their own particular risks (Table 57-3). The most common syndrome in older adults with ID is DS, and discussion of the various problems in people with DS can provide a general approach to such problems in other syndromes. Congenital heart disease is common in many ID syndromes as multiple genes contribute to its etiology; DS is a common cause of ID-associated congenital heart disease. Nowadays there is no reason why children with DS and congenital heart defects should not have surgical correction.26 Complications of uncorrected congenital heart disease, such as Eisenmenger syndrome and infective endocarditis, are thus becoming rare. In addition, persistent atrial septal defects are associated with increased risk of cerebral embolic events. However, many people with DS are not under regular follow-up once they become adults and may continue to have problems with arrhythmias. In particular, right bundle branch block is not uncommon after surgery. This is usually of no relevance until some form of left bundle branch block occurs when progression to complete heart block becomes more likely. There may also be residual hypoxemia due to persisting right-to-left shunts. Again, this generally is of no consequence until some extra stress to the system occurs, such as a general anesthetic. Some residual shunts may also be associated with a degree of pulmonary hypertension. Lifestyle factors and associated obesity put adults with DS at increased risk of hyperlipidemia. It is unclear how much impact this has on cardiovascular disease risk in this population, but there is evidence for a deleterious effect on cognition (see the next section). Dementia is three to four times more prevalent among adults with ID than the similarly aged general population.27 The prevalence in DS is substantially higher. Forty percent of those older than 50 years acquire an Alzheimer-type dementia with the typical neuropathologic features present in nearly everyone by the age of 40 years.28 Dementia incidence in those older than 50 is 18%,29 indicating the very short survival once the condition is diagnosed. Despite similar neuropathologic features, clinical manifestation of dementia often differs in DS with frontal lobe symptoms, such as deficits in executive functioning characterized by planning problems, personality changes, and development of problem behaviors being present at an early stage. It is natural to attribute any such changes to the onset of a dementing illness in DS because dementia is so common, but other conditions, even something as simple as constipation, can also occur with similar atypical symptoms. A full health assessment, with attention to physical factors, is therefore necessary. Epilepsy is common in ID, including DS, and may signal the onset of dementia in older adults; this may be especially the case for late-onset myoclonic epilepsy. People with more severe ID are at increased risk of seizures. Seizures are usually controllable with monotherapy. In other syndromes, seizures can be far more difficult to control. Visual problems in DS may be associated with development; approximately 60% of children with DS require glasses. People with DS have a flat nasal bridge, which can result in their glasses slipping. Strabismus is also common. In later life, cataracts are highly prevalent, nearly 30% in those aged 65 and older.30 Hearing impairment is also common and, similar to visual impairment, may date from childhood. Hearing impairment may result from a buildup of ear wax, but hearing aids are often needed for both conductive and sensorineural deficits.
Intellectual Disability in Older Adults
Definition and Causes
Syndrome Name
Chromosomal Abnormality
Phenotypic Appearance*
Angelman syndrome
15q11-q13 in the maternally contributed chromosome (cf. Prader-Willi syndrome has same deletion in paternal chromosome); a few cases due to paternal chromosome 15 disomy and a few due to putative single gene mutation on chromosome 15
Microcephaly, ataxic gait, strabismus, scoliosis
Down syndrome
Vast majority trisomy of chromosome 21; a few have trisomy 21 mosaicism; small proportion translocation of chromosome 21
Flat facial profile, epicanthic fold, relative hyperglossia, single palmar crease
Fragile X syndrome
Broad forehead with long face, large ears, strabismus, high arched palate, macroorchidism, scoliosis, joint hyperextensibility
Klinefelter syndrome
XXY and XXY mosaicism with variants XXXY or XXYY
Taller than average, microorchidism, youthful appearance, gynecomastia
Turner syndrome
XO, partial deletion of second X chromosome, XO mosaicism
Short stature, premature ovarian failure, high arched palate, low-set ears, webbed neck, strabismus, cubitus valgus, scoliosis, short fourth metacarpals
Williams syndrome
Deletion of CLIP2, GTF2I, GTF2IRD1, LIMK1, and other genes from chromosome 7
“Elfin” features of upturned nose, widely spaced eyes, wide mouth with full lips, small chin, high levels of empathy and anxiety
Epidemiology of Intellectual Disability and Aging
Prevalence
Incidence
Mortality
Biologic Aging in Individuals with Intellectual Disability: Syndromic and Nonsyndromic
Age-Related Disease: Syndromic and Nonsyndromic Patterns
Down Syndrome: Effects on Systems
Cardiovascular: Late Effects of Congenital Heart Disease, Hyperlipidemia
Neurologic: Dementia, Epilepsy, Vision and Hearing Loss
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Intellectual Disability in Older Adults
57