Fig. 9.1
The three main signaling pathways of UPR. UPR begins in the ER. GRP78 binds the main stress response proteins, IRE1α, ATF6, and PERK in an inhibitory fashion under normal conditions. When ER senses stress, GRP78 dissociates from the proteins under its charge to bind the unfolded proteins and chaperone them to be degraded, thus releasing and activating IRE1α, ATF6 and PERK. PERK dimerizes, autophosphorylating and facilitates the phosphorylation of eIF2a (this process can be inhibited by AKT as illustrated by the red arrow). A phosphorylated eIF2a proceeds to stop further protein translation, as well as activate the transcription factor ATF4. ATF4 enter the nucleus, binds DNA and promotes the gene expression of various proteins to increase the stress response, including CHOP, transport proteins, and proteins to decrease the amount of reactive oxidative species (ROS). When ATF6 dissociates from GRP78 it is still inactive and translocates to the Golgi apparatus where it is cleaved to its active form and then translocated into the nucleus and functions as a transcription factor, to promote gene expression of CHOP, additional GRP78, and XBP1 mRNA. The XBP1 mRNA exits the nucleus and is spliced by the active IRE1a into a shorter form, sXBP1 mRNA which is then expressed into the protein sXBP1. sXBP1(which can be inhibited through the activity of normal length XBP1 as illustrated) enters the nucleus as a transcription factor to promote the increased effectiveness of the stress response in a positive feedback loop including the synthesis of additional XBP1, GRP78, and other stress response proteins
9.3.1 IRE1α
IRE1α is a dual function protein with both kinase and endoribonuclease properties (Yoshida et al. 2001). While the IRE1α variant is ubiquitously expressed, the IRE1β variant is predominantly expressed in intestinal epithelial cells (Schroder and Kaufman 2006). As mentioned previously, the primary goal of the UPR is to resolve ER stress and maintain homeostasis. However, sustained stress can prompt UPR target genes to promote programmed cell death. During ER stress, IRE1α is dephosphorylated and its endoribonuclease activity is attenuated to prepare the cell for a pro-death outcome (Lin et al. 2007). IRE1α is capable of activating the apoptotic-signaling kinase 1 (ASK1) and Jun-N-terminal kinase (JNK) that promote apoptosis (Urano et al. 2000). On the other hand, the trans-autophosphorylated (active) role of IRE1α can be viewed in a pro-survival context. Active IRE1α promotes the splicing of a critical pro-survival gene called X-box binding protein 1(XBP1) (Yoshida et al. 2001). Splicing of XBP1 is considered “unconventional” due to the fact that the majority mRNA splicing is regulated by splicosomes within the nucleus, whereas XBP1 splicing is regulated through the endoribonuclease activity of IRE1α in the cytoplasm (Yoshida et al. 2001). During this splicing process, a 26 nucleotide intron is removed. This creates a frame-shift which encodes a larger version of XBP1 (sXBP1) which functions as a transcription factor (Feldman et al. 2005). sXBP1 translocates to the nucleus to activate transcription of cytoprotective genes two ways: (i) by its ability to activate specific cAMP response elements (CREs) (Clauss et al. 1996), and (ii) through ER stress response elements (ERSE1) (Oyadomari and Mori 2004). sXBP1 also upregulates the expression of GRP78, as well as additional XBP1 (Calfon et al. 2002). Research has also supported that the over-abundance of unspliced XBP1 actively works to suppress the activity of its spliced counterpart, which suggests that the relative balance between XBP1 and sXBP1 could be a significant factor in determining the ultimate fate of a cell (Lee et al. 2003).
XBP-1 may promote human breast carcinogenesis through impairment of cell differentiation regulation (Fujimoto et al. 2003). High levels of sXBP1 is associated with poor outcome (Davies et al. 2008) and antiestrogen resistance in estrogen receptor positive breast cancer (Gomez et al. 2007). Recently, sXBP1 expression has been associated with progression of triple-negative subtype, an aggressive form of breast cancer (Chen et al. 2014). Therefore, XBP1 is a potential target in anti-cancer drug development (Shajahan et al. 2009).
9.3.2 PERK
PERK contains a cytosolic protein kinase domain and is activated through trans-autophosphorylation and homodimerization. PERK activation inhibits further protein translation, thus, slowing down the influx of both folded and unfolded proteins into the ER adding to the stress (Harding et al. 2000). Activated PERK attenuates translation by phosphorylating eukaryotic translation initiation factor-2α (eIF2α) to prevent the assembly of the 80s-ribosome that is required to perform protein translation, therefore, halting the process of protein synthesis (Shi et al. 1998). Not all new protein translation is prevented since important cytoprotective UPR target genes and their transcription factors still need to be synthesized. To enable synthesis of these pro-survival proteins, phosphorylated eIF2α also selectively promotes the translation of specific mRNAs that possess small open reading frames on their 5′ untranslated region (Harding et al. 1999). Additionally, PERK allows the selective translation of specific proteins that can potentially resolve the ER stress, such as increasing p53 levels in the cell (Zhang et al. 2014), which is controlled by a ribosomal-Hdm2 that prevents the hdm2-mediated ubiquitination and degradation of p53. Increased levels of p53 allow greater control over the cell cycle, which in turn allows the cell to adequately repair and/or replicate its DNA properly. Furthermore, PERK mediated attenuation of translation can activate the avian reticuloendotheliosis virus-T leukemia (Rel) family transcription factors, including the pro-survial molecule, NF-kB (Schroder and Kaufman 2006).
Activating Transcription Factor 4 (ATF4) is another protein that is translated following PERK activation (Harding et al. 2000). Synthesized ATF4 enters the nucleus to induce gene expression of transcriptional regulators involved with various functions that will either help the cell relieve the ER stress and return the cell to homeostasis, or induce apoptosis. Such gene products produced include those involved in correcting the redox status of the cell, cellular metabolite synthesis and transport, and the pro-apoptotic protein C/EBP homologous protein, CHOP (also known as growth arrest- and DNA damage- inducible gene 153, GADD153) (Vattem and Wek 2004). CHOP is an important player in the induction of apoptosis when ER stress is unable to be resolved and will be discussed later in the chapter. Interestingly, PERK can be inhibited through AKT phosphorylation, which would prevent the phosphorylation of eIF2α, and thus shutting this branch of the UPR signaling cascade. Further investigation into this pathway has shown that inhibition of PERK in tumor cells can induce cell death (Mounir et al. 2011). Collectively, PERK and its downstream effector, eIF2α, fulfill a pro-survival role in UPR.
9.3.3 ATF6
Upon detection of ER stress, ATF6 is released from GRP78 and is translocated to the Golgi apparatus. There, it is cleaved by two proteases, separating its cytoplasmic domain with its membrane bound domain. There, the serine protease site-1 protease (S1P) cleaves ATF6 in its luminal domain. Consequently, metalloprotease site-2 protease (S2P) then cleaves ATF6 in the transmembrane domain and releases its bZIP transcription factor domain into the cytosol (Schroder and Kaufman 2006). The cleaved version of ATF6 is then translocated into the nucleus where it binds the ER stress response element CCAAT(N)9CCACG to initiate the gene expression of UPR target genes (Haze et al. 1999). Such target genes include GRP78, CHOP, and XBP1, in addition to other genes that code for proteins needed to help with protein folding, transport, and degradation (Okada et al. 2002).
9.4 Cell Death Mechanisms
Prolonged ER stress can lead to cellular suicide in order to protect the integrity of the system it belongs to. Thus, activation of UPR is closely connected to activation of programmed cell death pathways. Programed cell death is a complex and vital cellular function. These pro-death pathways are composed of several varied mechanisms which will be detailed below. The distinct hallmark of cancer includes the cellular ability to resist or subvert cell death (Hanahan and Weinberg 2000). Indeed, cancer cells acquire various mechanisms to escape or block the induction of programmed cell death. Large amounts of research have been carried out to investigate these cell death mechanisms to better understand these pathways in carcinogenesis. There are three main modes of programmed cell death: apoptosis, autophagy, and necrosis.
9.4.1 Apoptosis
Apoptosis (also known as Type I Programmed Cell Death) is a vital aspect of numerous processes, including cell turnover, immune system activity, embryonic development, and most relevant to cancer, chemical/radiation-induced cell death (Fig. 9.2). Apoptosis, an energy-dependent process, is characterized by loss of mitochondrial membrane potential, plasma membrane “blebbing, (bulging irregularly)” cell shrinkage, and nuclear fragmentation (Hotchkiss et al. 2009). There are many facets to the mechanisms of apoptosis, and expectantly, these mechanisms involve signaling cascades complete with sophisticated feedback responses. In cancer, apoptosis is particularly important and it is induced by therapeutic agents to reduce or eliminate malignant growths.
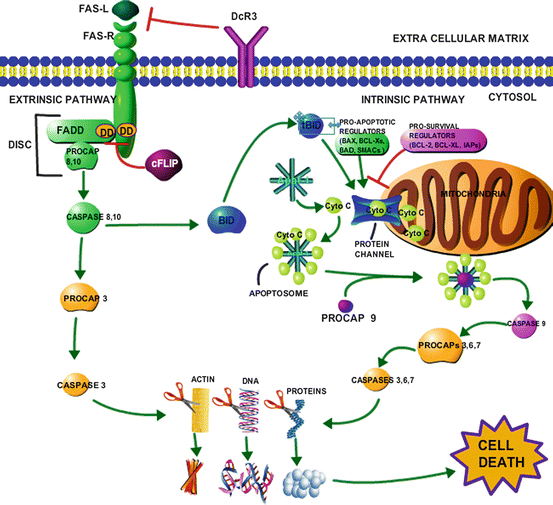
Fig. 9.2
Intrinsic and extrinsic apoptotic pathways and their convergence. The following shows the cascade of events that occur in each form of apoptosis. In the extrinsic pathway we use the FAS(R) receptor for an example, however, the same series of events occurs through the stimulation of other death receptors (TRAIL, TNF DR4/5) and their respective ligands and adaptors. Upon ligand binding, FADD docks with the receptor at the shared death domain (DD),thereby recruiting procaspase 8, 10 forming (DISC). Procaspase 8 and 10 are cleaved from FADD and activated into caspase 8, 10 (the initiator caspases). Caspase 8, 10 perform two functions: they cleave BID into a tBID which goes on to be used in the intrinsic pathways, and they cleave procaspase 3 into its active form caspase 3 (the executioner caspase). Caspase 3, which is also the executioner caspase in the intrinsic pathway proceeds to degrade many cellular materials including, actin in the cytoskeleton, DNA and other nuclear proteins, cytoplasmic proteins and organelles until the cell is so unstable that it causes a systematic failure and death. The extrinsic pathway has a few negative regulators illustrated with the red inhibition arrows; DcR3 competitively binds the FAS ligand to prevent the whole signaling cascade, as well cFLIP prevents the docking of FADD to the DD of the FAS receptor, preventing the activation of procaspase 8, 10. Within the intrinsic pathway, mitochondrial outer membrane permeabilization (MOMP), triggers the assembly of pro-apoptotic regulators, and the deactivation of pro-survival(apoptotic inhibiting) regulator to create protein channels that allow the release of cytochrome c (Ctyo C) from the intermembrane space. Apaf-1 binds cyto c to form the apoptosome (forming a wheel-like shape), which binds and activates procaspase 9 into caspase 9(initiator caspase). Procaspase 9 activates the executioner caspase 3, 6, 7 to fulfill the same role that they fulfill in the extrinsic pathway, thus causing cell death
9.4.2 The Intrinsic Pathway of Apoptosis
There are currently two main pathways in apoptosis: the intrinsic pathway and the extrinsic pathway. The intrinsic pathway is initiated when specific pro-apoptotic signals such as irreparable DNA damage or other serious stresses cause mitochondrial outer membrane permeabilization (MOMP), opening protein channels to allow the release of cytochrome c, a highly soluble heme protein and electron carrier found in between the mitochondria’s inner and outer membranes employed in the electron transport chain (Tafani et al. 2002). Cytochrome c forms a wheel-like complex with apoptotic protease activating factor-1(Apaf-1) creating the “apoptosome.” Procaspase 9, the initiator caspase (family of potent cysteine-dependent, aspartate-specific proteases), is bound by the apoptosome and modified to its enzymatically active form, caspase 9. Caspase 9 will then cleave the executioner procaspases, procaspase-3, 6, and 7, thus converting them to their active caspase form. These active executioner caspases cleave cellular substrates and catalyze the fragmentation and degradation of DNA, the cytoskeleton, and nuclear proteins, as well as the formation of apoptotic bodies, cross-linking of proteins, ligand expression for phagocytic cell receptors, and finally the uptake of degraded cellular material by phagocytic cells (Elmore 2007). The intrinsic apoptotic pathway is tightly regulated by a large family of proteins known as the B cell lymphoma 2 (Bcl-2) family which consists of at least 25 genes that contain up to 4 specific Bcl-2 Homology (BH1-4) domains that mediate hetero-and homodimerization with each other. This family consists of proteins with wide variability in function, an can be arranged based on how they regulate apoptosis; whether they perform a pro-apoptotic function (i.e. Bcl-Xs, Bax, Bak, Bim) or an anti-apoptotic function(i.e. Bcl-2, Bcl-XL, Bcl-w, Bfl-1). During homeostasis, these regulators, in tandem with other regulatory mitochondrial proteins known as small mitochondria-derived activators of caspases (SMACs) and their counterparts, inhibitors of apoptosis proteins (IAPs) (Fesik and Shi 2001), often hold each other in check. As a result, the fate of the cell is largely determined by the balance in the activation of these pro-death and pro-survival proteins (Karam et al. 2007). In many cancers that attempt to escape Bcl-2 mediated apoptosis, the pro-survival regulators will be overexpressed (Bcl-XL) and/or pro-death regulators (Bax) will be inhibited. The expression patterns of certain regulators can even help to predict the efficacy of certain treatments; for example, expression of Bcl-2 can predict the outcome of radiotherapy in laryngeal cancer with an accuracy of 71 % (Nix et al. 2005).
9.4.3 The Extrinsic Pathway of Apoptosis
The extrinsic pathway is initiated at the plasma membrane via the ligand binding and activation of cell surface death receptors (Ashkenazi and Dixit 1998). There are several death receptors (Fas, Trail-R1, TNFR), all with their own domain adaptors (FADD, TRADD) and ligands (FasL, Trail, TNFα); one main death receptor is Fas (also called FasR or apoptosis antigen 1) whose corresponding ligand and adaptor domain is FasL and Fas Associated Death Domain (FADD), respectively (Wajant 2002). These adaptors are recruited via their specific death domains to the receptor upon ligand binding and are necessary for facilitating the assembly of the Death Inducing Signaling Complex (DISC) which comprises procaspases 8 and 10, waiting to be cleaved and activated to then initiate the same execution pathway observed in the intrinsic pathway. In this pathway, FADD and DISC are homologous to Apaf-1 and the apoptosome from the intrinsic pathway respectively. Several decoy death receptors (DcRs), members of the tumor necrosis factor receptor (TNFR) superfamily can compete with the legitimate death receptors for ligand binding to avoid apoptosis (Zong et al. 2014). These decoy receptors are often overexpressed in cancer (Huang et al. 2014). Another inhibitory mechanism exists through the ability of cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein (cFLIP) to bind the death domain of FADD (and other adaptors), preventing the binding of the initiator pro-caspases 8 and 10, and thus inhibiting apoptosis (Salvesen and Walsh 2014).
The intrinsic and extrinsic pathways of apoptosis are linked. While both of these pathways are initiated uniquely, they both converge on the same execution pathway. Additionally, member of the Bcl-2 family of apoptotic regulators, BH3 Interacting Domain (Bid) function in both pathways. In the extrinsic pathway, when capsase 8 becomes activated, it cleaves cytoplasmic BID into a truncated form (tBid) which translocates to the mitochondria to help facilitate the release of cytochrome c to stimulate increased apoptosis via the intrinsic pathway (Brasacchio et al. 2014).
9.4.4 Autophagy
Autophagy (Type II Programmed Cell Death) is a cannibalistic process of self-degradation of cytoplasmic materials, translating quite literally to “self-eating” (Fig. 9.3). Autophagy is identified by the swelling of the vacuole to massive proportions within the cell, loss of organelles, and the appearance of many autophagosomes (Kroemer and Levine 2008). There are three types of autophagy: chaperone-mediated autophagy (CMA), microautophagy, and macroautophagy (the type that is most commonly referred to plainly as “autophagy”) (Jin et al. 2013). This process has long been thought to be a broad-scope, non-selective process, however, recently several forms of selective autophagy have been identified.
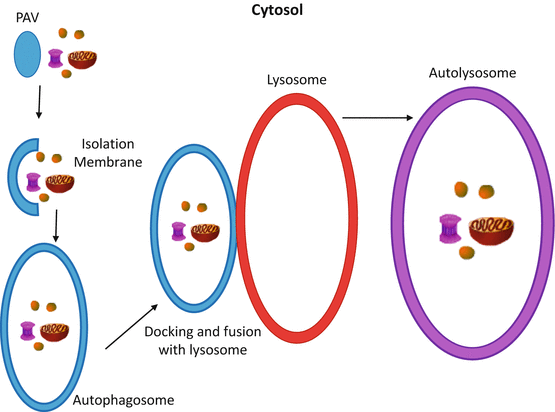
Fig. 9.3
The progression of autophagy. Autophagy begins with the pre-autophagic vesicle (PAV) and the cellular materials (i.e. misfolded proteins, ribosomes, mitochondria, etc.) that have been selected to be broken down into their raw materials. The PAV begins to elongate to create the isolation membrane, which will begin to engulf the targeted materials to eventually encase them in the autophagosome. The autophagosome will then traffic the cellular material to the lysosome (or vacuole; same process) where it will dock with the membrane of the lysosome. As this process continues, the autophagsome expels its contents into the lysosome and fuses with the membrane further, creating the autolysosome. The cellular materials are digested and reduced to their raw metabolites for use elsewhere in the cell
CMA is one such selective form of autophagy which applies most appropriately to UPR in that improperly folded proteins in the cytoplasm are systematically targeted and directly translocated into the lysosome to be degraded. This translocation is mediated by Heat Shock Cognate 70 (HSC70 aka HSPA8) as well as lysosome-associated membrane protein 2(LAMP2) (Dice 2007).
Microautophagy can be both selective and non-selective. Microautophagy describes the process by which the lysosome directly consumes and degrades cytoplasmic materials. Selective forms of this type include microautophagy of the mitochondria (micromitophagy), peroxisomes (micropexophagy), and the nucleus (Krick et al. 2008). While microautophagy and macroautophagy employ separate signaling pathways, some of the autophagy-related proteins are required in both processes (Li et al. 2012).
Macroautophagy (referred to hereafter simply as, autophagy) requires the formation of multi-membrane organelles known as autophagasomes to engulf cytoplasmic materials. These autophagasomes will transport the cellular material destined to be degraded to the lysosome whereby the autophagasome will fuse with and disperse its contents into the lysosome. While non-selective forms of autophagy exist, there are many forms of selective autophagy including: ribophagy (ribosomes), mitophagy (mitochondria), pexophagy(peroxisomes), glycophagy (glycogen), lipophagy (lipid droplets), xenophagy(foreign pathogens), zymophagy (secretory molecules), and perhaps most relevant to this chapter on UPR, ER-phagy. ER-phagy is induced to prevent the excessive distension of the ER during stress caused by the aggregation of unfolded proteins (Yorimitsu and Klionsky 2007). Indeed, researchers have observed the formation of autophagasomes simultaneously with the induction of UPR (Bernales et al. 2006). Interestingly, there has been research that shows that the degradation of cellular materials during autophagy occurs in a certain order; starting with cytosolic and proteosomal proteins proceeding further to organelles (Kristensen et al. 2008).
In general, the events that require the formation of autophagosomes, engulfing of cellular material, and docking to the lysosomes are controlled through the unimaginatively termed, autophagy-related proteins (ATGs), encoded via the ATG genes (Maiuri et al. 2007). The initiation of autophagy is controlled by ATG1, ATG13, and ATG17; the induction of this process requires a signal from the nutrient sensing protein, mTOR, which catalyzes the dephosphorylation of ATG13 which forms a complex with ATG1 and ATG17. The formation of the autophagosome proceeds through to vesicle nucleation and the formation of an isolation membrane controlled via ATG6 (aka Beclin-1). Once this occurs, mTOR and BCL-2 in the ER must be suppressed before ATG6 and ATG13 can form the autophagosome. The vesicle then elongates to consume target cytoplasmic materials; this is achieved through the action of several ATG proteins including: ATG3 ATG4, ATG5, ATG7, ATG10, ATG12, and ATG16. After the autophagosome has been assembled, the autophagosome will dock with the lysosome, fusing membranes and allowing the contents of the autophagosome to enter the lysosome to be degraded by lysosomal enzymes (Maiuri et al. 2007).
Autophagy is an important albeit unclear subject in the scope of cancer research. Researchers propose that autophagy fulfils a pro-survival role due to the necessity to overcome stresses such as nutrient depletion and hypoxia (Hanahan and Weinberg 2000). However, autophagy is a cell death mechanisms responsible for the extermination of cancer cells. Autophagy can have a profound effect on the efficacy of chemotherapeutic treatments and the development of resistance to anti-cancer therapies. When autophagy is inhibited, the therapeutic responses of resistant cancer cells to chemo-, endocrine- or radio-, therapies is increased, further supporting the pro-survival role of autophagy in cancer cells (Cook et al. 2011, 2012; Schwartz-Roberts et al. 2013). Autophagy is also instrumental in the cross-talk between the tumor-microenviroment and the cancer cells themselves, specifically in the syncing of metabolic activity (Pavlides et al. 2012).
Pro-survival autophagy is closely connected to the UPR. When the integrity of protein folding is compromised, and the continuous misfolding of proteins leads to energy depletion in the cell, UPR initiates and signals an up-regulation of autophagy in an attempt to recycle proteins to generate new metabolites and ATP, ideally alleviating the stress (Clarke et al. 2011, 2012; Cook et al. 2011, 2012). It is important to note that the nature of the autophagic response is not so dramatic as to be switched-on or switched-off, rather it is gradual; the level of autophagy increases or decreases to appropriate itself with the current level of stress (Tyson et al. 2011). However, what complicates autophagy in cancer is that although it is generally accepted as a pro-survival mechanism, excessive or prolonged stress will inevitably trigger cell death by arresting autophagy, which can in turn induce apoptosis or necrosis (Crawford et al. 2010; Schwartz-Roberts et al. 2013). An intricate relationship is emerging between autophagy and apoptosis, but unfortunately, there is little consensus among researchers. For instance, it is unclear whether what we describe as autophagic cell death is a truly novel form of cell death executed solely through autophagy, or if instead we are witnessing a tandem effort of autophagy and apoptosis in which the former simply initiates the latter. There are several hypotheses based on different cell lines and models. In the case of true autophagic cell death, the total volume of the autophagic vesicles would be equal or greater than the amount of free cytosol, thus a huge proportion of cytoplasmic material as well as organelles would be destroyed; this would cause irreversible damage to the cell, inducing atrophy and failure of cellular functions still intact leading inevitably to the death of the cell (Lum et al. 2005). In the scenario where autophagy triggers other forms of cell death, autophagy is first initiated as a response to stress stimuli, however, when the stress is not able to be resolved, apoptosis or necrosis is triggered (Espert et al. 2006).
9.4.5 Necrosis
Necrosis (Type III Programmed Cell Death) is a much less selective process, and is commonly associated with injury induced by physical trauma. Necrosis causes inflammation in groups of cells and is found commonly in the center of solid tumors. It is characterized by organelle and plasma membrane swelling, and rupture (Hotchkiss et al. 2009). It was long thought that necrosis was not regulated through signaling pathways and was only a passive form of cell death, however, research in the last decade has proved otherwise. Necroptosis, a programmed form of necrosis dependent on receptor-interacting protein kinase-3 (RIPK3), has been identified and is seen to be mediated in the cell through the binding of Tumor Necrosis Factor (TNF) to its cellular receptor (TNFR) which binds to and activates specific cell death receptors(Galluzzi and Kroemer 2008). Precise mechanism of the regulatory pathway of necroptosis remain largely undiscovered, however, recent research suggest that it is related to the other forms programmed cell death since necroptosis can be induced when both autophagy and apoptosis are blocked (White 2008).
9.5 UPR in Drug Resistant Cancer
As we have detailed above, UPR in a normal cell is a vital function for maintaining homeostasis and keeping the cell healthy. Unfortunately, cancer cells can take over this protective mechanism for their own survival, and to enable themselves to adapt to cellular stress and escape programmed cell death. As stated previously, hypoxia is a source of ER stress which is commonly found in solid tumors; UPR is commonly seen to be up-regulated in solid tumors, helping to relive the hypoxic stress and contributing to a poor clinical prognosis due to the tumor’s ability to withstand radio- and chemotherapy (Koumenis and Wouters 2006). UPR also helps cancer cells survive stresses such as genomic instability caused by a dysregulated cell cycle, nutrient and energy depletion, but most importantly in the clinical setting, DNA damaging anti-tumor therapeutics. Indeed, cancer that is successful in overhauling its UPR mechanism will become significantly more resistant to therapy (Clarke et al. 2012; Cook et al. 2012). Additionally, UPR can induce a quiescent-like state in which the cell growth is arrested, yet survival remains competent, to allow them to resist drug/radiation induced stress (Ranganathan et al. 2006).
There has been a great deal of research hinged on understanding this form of acquired resistance in tumors, and more importantly, finding ways to circumvent the resistance to restore sensitivity to therapeutics. In research on hepatocellular carcinoma (HCC), a particular cancer biomarker, CD147, was identified as an inducer of UPR closely associated with the expression of GRP78 (which is also seen to be overexpressed in many malignant diseases (Cook et al. 2012). When CD147 was up-regulated, apoptosis in HCC was inhibited, and a decreased sensitivity to Adriamycin (chemotherapeutic agent) was observed; thus CD147 (or other UPR-related proteins) inhibition presents itself as an encouraging opportunity to improve the efficacy of anti-cancer therapeutics (Tang et al. 2012). In human epidermoid carcinoma (HEp3) cells, cell cycle regulator p38 plays a role in the activation of PERK and up-regulation of GRP78; moreover the latter is required to facilitate the inhibition of Bax (a pro-death regulator of apoptosis) activation to promote cell survival and drug resistance (Ranganathan et al. 2006). Further research into the role of GRP78 in cancer treatment showed that the forced overexpression of GRP78 conferred resistance to topoisomerase inhibitors (apoptosis inducers) across various tumor cell types (Reddy et al. 2003).
9.6 Conclusions and Future Perspectives
The UPR is a highly conserved adaptive mechanism that is instrumental in maintaining cellular homeostasis in response to ER stress in normal cells. In cancer, up-regulation of the UPR can protect the cell from ER stress resulting from therapeutic interventions. While the precise mechanisms of the UPR activation in cancer development remains unknown, it is clear that targeting pro-survival players in the UPR pathway can help promote cell death in various cancer cell and tumor models. Investigating novel therapeutic targets in the UPR pathway is an active area of research in cancer (Table 9.1). As we understand the complexity of the UPR and the programmed cell death pathways, it is likely that combination therapies that induce pro-death but inhibit the pro-survival pathways of the UPR and autophagy will be most effective to inhibit survival in cancer cells. Moreover, the activation of specific arms of the UPR is likely to be dependent on the cellular context, and therefore, knowledge of the signaling mechanism associated with the specific cancer type is needed. In summary, the UPR is a promising yet perplexing area of cancer research. More research is needed to understand the role of the resilience of UPR in averting cell death and promoting cancer growth in human body.
Table 9.1
 Introduction to Cell Stress Responses in Cancer: The Big Picture
Introduction to Cell Stress Responses in Cancer: The Big Picture
 Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
 Inflammatory Dysregulation and Cancer: From Molecular Mechanisms to Therapeutic Opportunities
Inflammatory Dysregulation and Cancer: From Molecular Mechanisms to Therapeutic Opportunities
 Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
 The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
 Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells
Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells




Drugs (both pre-clinical and clinical) targeting UPR and cell death pathways
Related posts:
Introduction to Cell Stress Responses in Cancer: The Big Picture
Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
Inflammatory Dysregulation and Cancer: From Molecular Mechanisms to Therapeutic Opportunities
Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
