Myelodysplastic syndromes (MDS) are heterogeneous clonal hematologic malignancies characterized by cytopenias caused by ineffective hematopoiesis and propensity to progress to acute myeloid leukemia. Innate immunity provides immediate protection against pathogens by coordinating activation of signaling pathways in immune cells. Given the prominent role of the innate immune pathway in regulating hematopoiesis, it is not surprising that aberrant signaling of this pathway is associated with hematologic malignancies. Increased activation of the innate immune pathway may contribute to dysregulated hematopoiesis, dysplasia, and clonal expansion in myelodysplastic syndromes.
The innate immune system is an evolutionarily conserved defense mechanism against pathogens. Unlike the adaptive immune response, the innate immune system does not recognize every foreign pathogen, but instead recognizes related components shared by many microbes, referred to as “pathogen-associated molecular patterns.” Coordinated activation of the innate immune system is achieved by key sentinel cells and a complex sequence of events, including engagement of pathogen-associated molecular patterns responsive receptors, recruitment of phagocytic cells, release of inflammatory mediators, and activation of the complement system resulting in pathogen clearance. The toll-like receptor (TLR) family plays a major role in the initial detection and subsequent elimination of foreign pathogens. This process is achieved through activation of intracellular signaling pathways, such as NF-κB and MAPK, which initiate a coordinated set of responses.
The innate immune signaling complex is activated by functionally related receptors
Activation of the innate immunity pathway is the result of the interaction between a ligand (microbial product) and its cognate receptor. The human TLR family comprises 10 members (TLR1–TLR10) involved in the recognition of microbial products, such as lipopolysaccharide (LPS), lipoteichoic acid, peptidoglycan, microbial lipoproteins, and viral nucleic acids (DNA and RNA). The TLRs exhibit homology to the interleukin-1 receptors (IL-1R) and tumor necrosis factor receptors (TNFR). IL-1Rs are a functionally related family of receptors that rely on TRAFs (TNF associated factors) for transmitting intracellular signals. Despite the functional similarities to TLRs, IL-1Rs are not primary sensors of infection but rather secondary sensors that respond to IL-1 from sentinel cells, such as macrophages that have been previously infected. The primary role of IL-1Rs is the production of proinflammatory genes. The TNFR members (eg, TNFR1, LT-βR, Fas, CD40, RANK, EDAR, and LMP) also functionally overlap with innate immune receptors. The main purpose of the TNFR family is to activate genes involved in inflammation and immunoregulatory responses, cell proliferation, viral responses, and growth inhibition. Ligands responsible for activation of TNFR include TNF-α, lymphotoxin (LT), Fas ligand, CD40 ligand, RANK ligand, and EDA. Research on the IL-1R and TNFR family members has revealed mechanistic similarities and related components shared with the TLRs.
Components of the innate immune signaling system
The best-described TLR member, TLR4, is the receptor for LPS. Two pathways diverge downstream of TLR4, the myeloid differentiation primary response gene 88 (MyD88)-dependent and -independent pathways, resulting in the expression of inflammatory cytokines or interferon (IFN)-inducible genes, respectively ( Fig. 1 ). The MyD88-dependent pathway mediates a rapid and acute response, whereas the MyD88-independent pathway is responsible for a delayed response. On LPS recognition, a complex is formed on the outer plasma membrane consisting of LPS, TLR4, MD2, and CD14. The LPS-TLR4 interaction is stabilized by MD2 and CD14 coreceptors, which then facilitates intracellular recruitment of protein adaptors required for downstream signaling. MyD88 is a toll–interleukin 1 receptor (TIR)-containing protein adaptor that forms a complex on the corresponding intracellular TIR domain of TLR4. Along with Mal/TIR domain–containing adaptor protein (TIRAP), MyD88 recruits interleukin-1 receptor-associated kinase (IRAK4), which then recruits IRAK1, resulting in subsequent autophosphorylation and disassociation from the TLR4-MyD88 complex. The MyD88-IRAK complex then binds to TRAFs, key effectors of the innate immune signaling complex. The TRAF-containing complex forms the key signaling component, which includes two kinase subunits, IKKα and IKKβ (inhibitor of NF-κB kinase alpha and beta), and a regulatory subunit (IKKγ/NEMO). One of the main TRAF members, TRAF6, is an E3 ligase that self-activates by autoubiquitination and also forms polyubiquitin lysine-63 chains on IKKγ resulting in IKK-mediated activation of NF-κB. Activation of the IKK complex by TRAF6 relies on another complex consisting of TAK1 (transforming growth factor-β activated kinase 1) and TAB1/2 (TAK1 binding protein-1 and -2). Engagement of the TAK1/TAB1/2 complex with TRAF6 not only induces NF-κB, but can also activate specific MAPK pathways (JNK and p38). In contrast, the MyD88-independent pathway forms a TLR4 complex containing TRIF and TRAM adaptor molecules (see Fig. 1 ). As in the MyD88-dependent pathway, TRAF6 plays a key role in downstream activation, but the kinases RIP1 and TBK1 are also necessary for NF-κB and IRF3 activation.
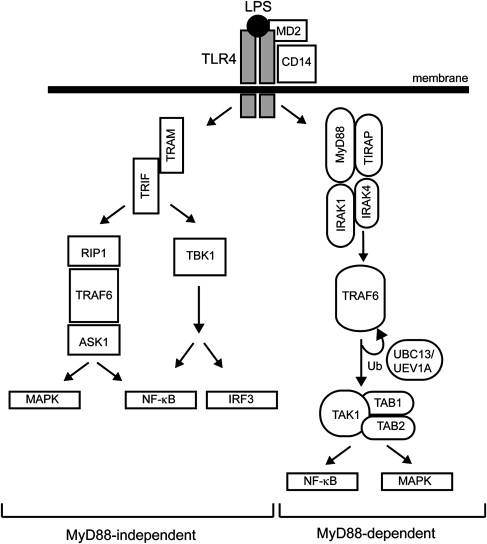
Components of the innate immune signaling system
The best-described TLR member, TLR4, is the receptor for LPS. Two pathways diverge downstream of TLR4, the myeloid differentiation primary response gene 88 (MyD88)-dependent and -independent pathways, resulting in the expression of inflammatory cytokines or interferon (IFN)-inducible genes, respectively ( Fig. 1 ). The MyD88-dependent pathway mediates a rapid and acute response, whereas the MyD88-independent pathway is responsible for a delayed response. On LPS recognition, a complex is formed on the outer plasma membrane consisting of LPS, TLR4, MD2, and CD14. The LPS-TLR4 interaction is stabilized by MD2 and CD14 coreceptors, which then facilitates intracellular recruitment of protein adaptors required for downstream signaling. MyD88 is a toll–interleukin 1 receptor (TIR)-containing protein adaptor that forms a complex on the corresponding intracellular TIR domain of TLR4. Along with Mal/TIR domain–containing adaptor protein (TIRAP), MyD88 recruits interleukin-1 receptor-associated kinase (IRAK4), which then recruits IRAK1, resulting in subsequent autophosphorylation and disassociation from the TLR4-MyD88 complex. The MyD88-IRAK complex then binds to TRAFs, key effectors of the innate immune signaling complex. The TRAF-containing complex forms the key signaling component, which includes two kinase subunits, IKKα and IKKβ (inhibitor of NF-κB kinase alpha and beta), and a regulatory subunit (IKKγ/NEMO). One of the main TRAF members, TRAF6, is an E3 ligase that self-activates by autoubiquitination and also forms polyubiquitin lysine-63 chains on IKKγ resulting in IKK-mediated activation of NF-κB. Activation of the IKK complex by TRAF6 relies on another complex consisting of TAK1 (transforming growth factor-β activated kinase 1) and TAB1/2 (TAK1 binding protein-1 and -2). Engagement of the TAK1/TAB1/2 complex with TRAF6 not only induces NF-κB, but can also activate specific MAPK pathways (JNK and p38). In contrast, the MyD88-independent pathway forms a TLR4 complex containing TRIF and TRAM adaptor molecules (see Fig. 1 ). As in the MyD88-dependent pathway, TRAF6 plays a key role in downstream activation, but the kinases RIP1 and TBK1 are also necessary for NF-κB and IRF3 activation.
Genes regulated by innate immune signaling
Over 1000 protein-coding genes are activated following stimulation of the innate immune system. The primary goal of the innate immune system is to generate an immune response by activating the inflammatory cascade. The best characterized genes directly regulated by the innate immune pathway include cytokines (IL-1β, IL-6, TNF-α, granulocyte colony–stimulating factor, granulocyte-macrophage colony–stimulating factor, and macrophage colony–stimulating factor); chemokines (membrane cofactor protein-1 and IL-8); and IFNs (IFN-β and IRG). More recent attention has focused on the regulation of noncoding RNAs, such as microRNAs (miRNAs), by the innate immune pathway. miRNAs are 21- to 25-nucleotide noncoding RNAs that posttranscriptionally repress specific mRNA targets through 3′-untranslated region interactions. At least five miRNAs (miRs) have been reported to increase in expression following stimulation with LPS. Four independent groups identified miR-146, miR-147, miR-155, miR-181, and let-7 as effectors of the TLR4 pathway. The precise role of these miRs in mediating the LPS-TLR4 response remains to be delineated. Mouse experiments have revealed, however, that miR-155 may be involved in driving myeloid expansion in vivo, and overexpression of miR-147 abrogates a macrophage inflammatory response as indicated by lower expression of TNF-α and IL-6. The roles of miR-146, miR-181, and let-7 have yet to be studied in a similar context, so it is unclear how they contribute to functional innate immune responses. There is some evidence that these miRNAs are important in hematopoiesis and may be involved in differentiation or survival of key immune cells. Knockdown of miR-146 in human or mouse hematopoietic progenitor cells results in increased megakaryopoiesis. Ectopic expression of miR-181 in hematopoietic stem and progenitor cells leads to B-lineage differentiation. Some of these miRNAs potentially function as negative-feedback regulators. For instance, miR-146 has been shown to target TRAF6 and IRAK1, and let-7 and miR-181 are predicted to target TLR4. It is conceivable that induction of these miRNAs dampens the LPS response by binding and inhibiting multiple components of the innate immune pathway ( Table 1 ).
| microRNA | Innate Immunity mRNA Target |
|---|---|
| miR-145 | TIRAP |
| miR-146 | TRAF6, IRAK1 |
| miR-147 | TLR4 |
| Let-7a | TLR4 |
| miR-181 | TLR2/4/8, NOD2, CARD8 |
Role of innate immune signaling on normal hematopoiesis
Hematopoietic Response to Acute Inflammation and Innate Immune Signaling Activation
Genetic and functional studies have revealed the consequences of innate immune signaling pathways on the distinct steps of hematopoietic differentiation following inflammation or pathogen infection. Extensive discussion and experimental evidence on this topic has been previously reviewed, so only a brief overview is provided. Inflammation and subsequent activation of innate immune signaling results in activation and expansion of mature leukocytes, such as monocytes, dendritic cells, macrophages, and neutrophils, but reduction of B lymphopoiesis in the bone marrow. Similarly in mice, administration of LPS through intravenous injection results in rapid and dynamic effects on myeloid and lymphoid populations in the bone marrow and blood. After LPS injection, expansion of granulocyte-macrophage populations and reduction in B cells and erythroid precursors are observed in the bone marrow by 72 hours. The inflammatory-mediated imbalance in lymphopoiesis and granulopoiesis is driven by growth factors, chemokines, and cytokines.
Role of Innate Immune Signaling in Hematopoietic Stem and Progenitor Cells
Most work has described the TLRs and the innate immune signaling pathway as critical responders to foreign pathogens in mature myeloid and lymphoid cells. Emerging evidence now suggests that TLR are also important signaling transducers in hematopoietic stem and progenitor cells. Functional TLRs and their coreceptors are expressed on multipotent hematopoietic stem cells. By binding to TLR4, LPS drives hematopoietic stem and progenitor cells to proliferate and differentiate into mature monocytes and macrophages at the expense of lymphoid differentiation in vitro ( Fig. 2 A). Similarly, mice administered LPS show evidence of activated hematopoietic stem cells in the bone marrow and preferential stimulation of macrophage and monocyte development, whereas B and T lymphoid production is impaired. These observations demonstrate a critical role of innate immune signaling in normal hematopoietic stem and progenitor cell differentiation.
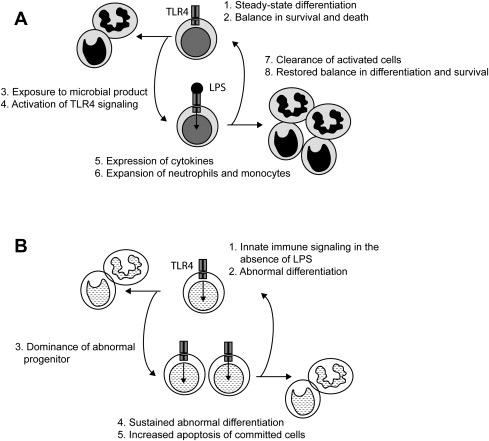
The importance of TLR4 signaling in hematopoietic stem cells is further supported by genetic experiments with TAK1, a member of the MAPKKK family and a mediator of TRAF6 signaling (see Fig. 1 ). Under normal physiologic conditions, TAK1 is expressed and activated in hematopoietic stem cells. TAK1 gene-targeted mice show ineffective hematopoiesis leading to pancytopenia. Notably, TAK1-deficient mice display reduced white blood cells, platelets, and nucleated cells in the bone marrow, but hemoglobin levels are unaffected. Detailed examination of the marrow indicates that depletion of TAK1 results in loss of primitive hematopoietic cells in the marrow. The loss of hematopoietic stem and progenitor cells in TAK1-deficient mice is secondary to increased apoptotic signaling, mediated through loss of NF-κB and JNK activation. TAK1 is a key gene of the innate immune pathway responsible for maintenance of hematopoietic stem cells.
Role of innate immune signaling in malignant hematopoiesis
Innate Immune Signaling Defect in Acute Myeloid Leukemia
Acute myeloid leukemia (AML) results from genetic defects in a hematopoietic stem and progenitor cell or myeloid lineage precursor. A combination of increased cell proliferation and a block in hematopoietic differentiation are hallmark criteria for the development of AML. Classification of AML assumes morphologic similarities of the leukemic cells to the normal cell counterpart within a particular stage of myeloid differentiation. Distinct recurring mutations have been identified in AML and are thought to be responsible for the defects associated with proliferation or differentiation of leukemic cells. Balanced translocations, such as t(15;17)/PML-RARA, t(8;21)/AML1-ETO, t(16;16)/CBFB-MY11, t(9;11)/MLLT3-MLL, or other rearrangements of 11q23/MLL are common in de novo AML. These alterations, which are now part of the World Health Organization classification of myeloid neoplasias, have been extensively characterized to reveal the roles of the fusion genes in leukemogenesis. In addition to genomic alterations, point mutations have also been identified. The most common mutations in AML are ones in the FMS-like tyrosine kinase 3 (FLT3) gene, CEBPA, and NPM1. Internal tandem repeats of FLT3 and overexpression of MN1 are also prognostically important in AML and have been shown to directly contribute to leukemogenesis in mouse models. Identification of these alterations in AML patients has improved diagnosis and treatment; however, the heterogeneity of AML and variable survival outcomes suggests that yet undiscovered genes and pathways contribute to AML.
The role of innate immune signaling in AML is becoming increasingly apparent, but the evidence is indirect. IL-1β has been shown to maintain survival and proliferation of AML blasts. Protein levels of TRAF family members are also increased in AML cell lines, but are low or undetectable in normal hematopoietic cells. Consistent with this observation, the downstream pathways regulated by TRAFs, such as NF-κB, are activated in AML blasts but not in normal CD34 + cells. The importance of NF-κB activation in AML is exemplified by studies using genetic and pharmacologic inhibitors of NF-κB. For example, inhibiting NF-κB signaling suppresses growth of AML blasts and leukemic stem cells.
The contribution of TNFR family members to hematologic malignancies is variable and dependent on the specific receptor. TNFR superfamily members that directly associate with Fas-associated death domain activate the extrinsic apoptotic pathway and have been reported to suppress growth of leukemic cells. In contrast, TNFR members that associate with TNFR1-associated death domain activate prosurvival signaling in parallel with apoptotic pathways, and facilitate leukemic cell growth. One TNFR-family member, CD30 variant, has been identified in a significant proportion (approximately 70%) of AML-M4 and M5. CD30 variant is thought to induce cell growth and differentiation through interactions with TRAF2 or TRAF5 proteins and subsequent activation of NF-κB in AML cell lines. The role of CD40 in AML is more confusing. CD40, which also belongs to the TNFR family, requires TRAFs to propagate signals to NF-κB, STATs, and MAPK. Despite the unresolved role of CD40, it is clear that the ligand for CD40 promotes proliferation, self-renewal, and increased survival potential of AML blasts. Furthermore, circulating levels of CD40 are elevated in AML patients and are associated with poor overall survival.
Individual components of the innate immune pathway have not been as extensively described in AML. Most of the reported research has described components of the MyD88-independent pathway. ASK1, a MAPK member of the MyD88-independent signal, is activated by TRAF6 in response to reactive oxidative species. Knockdown of ASK1 sensitizes AML cells to arsenic trioxide-mediated apoptosis. In contrast, azinomycin epozide induces apoptosis in AML cells through activation of ASK1 and caspase 3. The latter observation supports the original finding that ASK1 induces apoptosis by activating p38. The conflicting data on the role of ASK1 in AML may be resolved by examining ASK1 in a cell-context manner. For example, ASK1 may promote prosurvival pathways in primitive hematopoietic cells, but cell death in committed myeloid progenitors. That the MyD88-independent pathway is responsible for inducing apoptotic pathways is supported by a similar proapoptotic role of RIP1 in myeloid blast cell differentiation.
More recent evidence implicating a key role for innate immune signaling in AML was provided in a miRNA expression study on cytogenetically normal AML patient samples. Only 12 of more than 300 miRNAs examined correlated with overall survival. Five of these miRNAs belong to the miR-181 family. Increased expression of miR-181 family members was associated with decreased risk of death caused by AML. Furthermore, because miRNAs are negative regulators of specific mRNA targets, this study investigated genes and proteins that were inversely correlated with miR-181 expression in the patient samples. It was revealed that genes encoding TLRs (TLR2, TLR4, and TLR8), IL-1β, and various effectors of these pathways (eg, CARD8 and NOD2) were increased in patients with low miR-181 expression. Of these genes, TLR4, IL-1β, and CARD8 are predicted targets of miR-181. It is hypothesized that reduced levels of miR-181 result in increased signaling of the TLR4 or IL1R pathways and contribute to AML progression. In related studies, miR-181 family members have been associated with morphologic subclasses of AML and differentiation of hematopoietic precursors.
Sequential acquisition of genomic alterations, somatic mutations, and deregulation of key signaling pathways contribute to the transformation of a normal hematopoietic stem and progenitor cell to an AML blast. The critical events result in a block in differentiation and increased proliferation of myeloid precursors. According to what is known about signaling pathways downstream of TLRs, deregulation of innate immune signaling likely contributes to resistance to proapoptotic signals and increased survival of AML blasts. At this time, there is no evidence to suggest that innate immune signaling mediates a block in differentiation or promotes self-renewal of primitive hematopoietic stem and precursors resulting in AML.
Innate Immune Signaling Defects in Myelodysplastic Syndromes
The myelodysplastic syndromes (MDS) are a heterogeneous group of clonal hematologic malignancies characterized by peripheral cytopenias, a hypercellular marrow with ineffective hematopoiesis, and a propensity to progress to AML. MDS is thought to arise from a primitive hematopoietic progenitor that has acquired genetic or epigenetic abnormalities. The current World Health Organization classification of MDS comprises eight subtypes, based on biologic, genetic, and morphologic features. Activation of the innate immune system by chronic infection can reproduce hematologic abnormalities resembling MDS, including anemia, neutropenia, thrombocytopenia, and trilineage dysplasia. As examples, chronic infection with Leishmania or parvovirus have been reported to induce pancytopenia and dysplastic erythroid and myeloid precursors in the marrow. Other evidence that the innate immune system plays a role in MDS comes from the use of immune modulatory drugs for treatment of MDS. The best-known immune modulatory drug, lenalidomide, has been successfully used for treatment of MDS subtypes associated with deletion of chromosome arm 5q. These observations provide indirect evidence that activation of the innate immune pathway may mediate features of MDS.
There are few described mouse models that exhibit features of MDS. As evidenced by studies in AML, identification of genetic alterations and the creation of representative mouse model systems may provide insight into the biologic mechanism and potential therapeutic targets for MDS. To further the understanding of the biologic basis of MDS and identify novel therapeutics, efforts to unravel the genetic networks associated with MDS need to be undertaken.
Evidence of Direct Activation of Innate Immune Pathway Genes in MDS
We recently focused on candidate genes and their contribution to 5q syndrome, a common subtype of MDS. 5q syndrome originates in a hematopoietic stem cell and is defined by a heterozygous interstitial deletion of chromosome arm 5q, macrocytic anemia, neutropenia, and thrombocytosis associated with abnormal megakaryocytes. Although deletion of chromosome arm 5q was first reported over 30 years ago, the genes responsible for the clinical manifestation of this disease remained unknown until recently. miRNAs encoded on chromosome 5q were investigated as possible mediators of 5q syndrome. Deletion of chromosome 5q in MDS patients results in loss of at least two functionally related miRNAs (miR-145 and miR-146a). Based on miRNA target prediction algorithms and in vitro validation experiments, TIRAP was identified as a target of miR-145, and TRAF6 was confirmed to be a target of miR-146a (see Tables 1 and 2 ). TIRAP and TRAF6 are two proteins that lie in the MyD88-dependent pathway of innate immune signaling (see Fig. 1 ). Loss of either of these two miRNAs results in depression of innate immune signaling. Expression of TIRAP and TRAF6 proteins is relatively low in primitive hematopoietic cells; however, reduction in miR-145 and miR-146a levels during differentiation results in increased TIRAP and TRAF6 protein, respectively. This suggests that innate immune genes, such as TIRAP and TRAF6, are tightly regulated in hematopoietic stem and progenitor cells. Murine marrow transplant models in which miR-145 and miR-146a are reduced or TRAF6 is overexpressed results in megakaryocytic and platelet defects, and a propensity to develop acute leukemia or marrow failure, through both cell autonomous and nonautonomous mechanisms. Loss of miR-145 and miR-146a does not result in macrocytic anemia, a key clinical finding in 5q- syndrome. Identification of the RPS14 gene in the minimally deleted region of 5q- syndrome, however, likely explains the macrocytic anemia. The full spectrum of clinical features in 5q syndrome may be explained by the collective loss of miR-145, miR-146a, and RPS14.
Related posts:
 Epigenetic Changes in the Myelodysplastic Syndrome
Epigenetic Changes in the Myelodysplastic Syndrome
 Mouse Models of Myelodysplastic Syndromes
Practical Recommendations for Hypomethylating Agent Therapy of Patients With Myelodysplastic Syndromes
Mouse Models of Myelodysplastic Syndromes
Practical Recommendations for Hypomethylating Agent Therapy of Patients With Myelodysplastic Syndromes
 Prognostic Classification and Risk Assessment in Myelodysplastic Syndromes
Prognostic Classification and Risk Assessment in Myelodysplastic Syndromes
 Myelodysplastic Syndromes Classification and Risk Stratification
Myelodysplastic Syndromes Classification and Risk Stratification
 Patient Selection for Transplantation in the Myelodysp lastic Syndromes
Patient Selection for Transplantation in the Myelodysp lastic Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


