Inherited Dyslipoproteinemias of Various Etiologies
Familial lipoprotein(a) hyperlipoproteinemia
High levels of lipoprotein(a) (Lp(a)) aggregate or segregate in families and may be associated with atherosclerotic vascular disease. In Turkey, a country with a low average low-density lipoprotein-cholesterol (LDL-C) level, familial Lp(a) hyperlipoproteinemia has been reported as one of the most common familial lipoprotein disorder leading to premature myocardial infarction.
Lp(a) or lipoprotein(a) was first described in 1963 by Berg at the University of Oslo. This peculiar lipoprotein has a ‘recent’ evolutionary history as it is present only in humans, Old World primates and hedgehogs. Essentially, it consists of a typical LDL particle linked by a disulphide bridge to apolipoprotein(a). The latter is a large glycoprotein with no amphipathic helices (lipid-binding sites), which is linked to apolipoprotein B (apoB)-100 by a single disulphide bond in the C-terminal regions of both proteins, close to the LDL receptor binding site of apoB (4.1). In the early years of lipoprotein identification, Lp(a) could be demonstrated in plasma using a combination of lipoprotein electrophoresis and preparative ultracentrifugation. It was recovered in the bottom d = 1.006 g/ml ultracentrifugal fraction with high-density lipoproteins (HDL) and was given the name ‘sinking preb-lipoprotein’ (4.2). In 1987, McLean and colleagues found that both the apo(a) and the plasminogen genes had coding sequences for so-called kringle domains, K I to K V for plasminogen and K IV and K V for apo(a) (4.3). The hedgehog apo(a) has K III repeats. The structure of a typical kringle is shown in 4.4.
The apolipoprotein(a) gene (LPA, OMIM No. 152200) maps to chromosome 6q26-27 and is heterogeneous because of the number of sequence repeats (3 to >40) that encode the K IV type 2 domains. The inheritance pattern is complicated by the fact that the apo(a) size polymorphism is controlled by a large number of co-dominant alleles (>35) that vary widely in expressivity (4.5). The frequency distribution of kringle IV type 2 repeats and of Lp(a) plasma concentration differs widely across populations (4.6). The encoded apo(a) protein thus can have a molecular weight ranging from 300 kDa to 800 kDa. There is a general inverse relationship, with inter-individual variation, between the size of apo(a) and the plasma concentration of Lp(a), and this is believed to be 90% genetically determined. Most individuals are heterozygous for apo(a) size, therefore the plasma concentration of Lp(a) results from the relative contribution of each isoform. It has been observed that black people have two- to three-fold higher levels of Lp(a) than white people or Asians, even when adjusted for apo(a) size. However, an increased risk for coronary heart disease (CHD) associated with higher Lp(a) levels in African-Americans has not been documented. Interestingly, mutations of the apo(a) gene (4.7) resulting in no circulating apo(a) (Lp(a) deficiency) are associated with an apparently normal phenotype.
Very little is known about Lp(a) metabolism. However, it has been established that the rate of Lp(a) synthesis in the liver determines its plasma concentration. Lp(a) is assembled extracellularly by a two-step mechanism involving a conformational change and its catabolism is not affected by LDL receptor activity. Studies in an apoE-deficient subject and controls (see 3.35 and 3.36) have revealed that a buoyant form of Lp(a) associated with very-low-density lipoproteins (VLDL) and intermediate-density lipoproteins (IDL) may be a precursor of mature Lp(a) which does not need apoE to be cleared from plasma. Recent stable isotope studies indicate that in the fed state some apo(a) may dissociate from apoB and reassociate with another newly secreted apoB particle. Peptide fragments of apo(a) have been found in the urine. Lp(a) is an acute phase reactant and increases markedly in plasma in response to the inflammatory cytokine interleukin (IL)-6. Beside the implication in athero-thrombogenesis, the physiological function of apo(a) is not fully understood. Some of the structure-function relationships that have been established are described in 4.7.
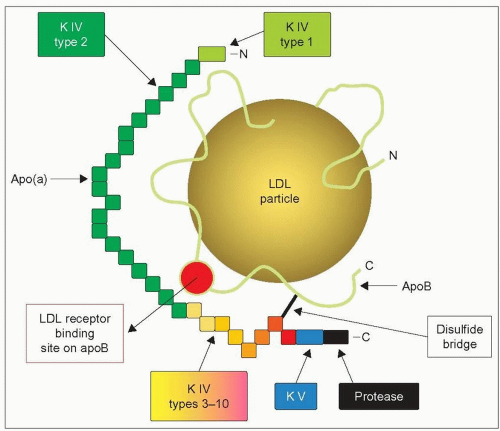 4.1 Model structure of lipoprotein(a) (Lp(a)). The first component of Lp(a) is similar to a low-density lipoprotein (LDL) particle consisting of a lipid core of cholesteryl esters and triglycerides surrounded by a surface layer of phospholipid and free cholesterol. Its single molecule of surface apolipoprotein B (apoB represented by a green-yellow line), is linked to its second component, apolipoprotein(a) through a single disulphide bond (black line). The putative LDL receptor binding domain of apoB is shown as a red circle. The apo(a) moiety consists of a single copy of kringle (K) IV type 1 (light green), kringle IV types 3-10 (yellow to orange), kringle V (blue), and a protease domain analogous to plasminogen (black). In addition, it contains multiple copies (3->40) of kringle IV type 2 (green) which are responsible for the size polymorphism of Lp(a). Kringles contain 80-85 amino acids for a molecular weight of about 10 kDa and so the molecular weight of apo(a) may range from 300 kDa to 800 kDa, depending on the number of kringle IV type 2 that are repeated. Note that apo(a) location is such that it might interfere in the interaction between apoB and its receptor. This diagram is redrawn and slightly modified from Berglund L, Ramakrishnan R (2004). Lipoprotein(a) An elusive cardiovascular risk factor. Arterioscler Thromb Vasc Biol, 24: 2219-2226. |
Many clinical studies, carried out mostly in white populations, have found significant correlations between the plasma level of Lp(a), and the risk for a variety of vascular diseases including coronary artery disease (CAD), peripheral vascular disease, vein graft stenosis after coronary artery bypass surgery, stroke, dementia and deep vein thrombosis. Plasma Lp(a) is elevated in patients with abdominal aortic aneurysm, systemic sclerosis, hypothyroidism, Behçet’s syndrome (a multisystem inflammatory disease with skin and mucosal lesions involving eyes, mouth and genitals, complicated by arthritis, thrombophlebitis and vascular events), gangrenous diabetic foot lesions, hemodialysis or end-stage renal disease. In the latter, Lp(a) is independently associated with cardiovascular events. A recent meta-analysis of 27 prospective studies cumulating in 5436 deaths from CAD or non-fatal myocardial infarction over a mean follow-up of 10 years yielded a significant 70% increase of cardiovascular risk for individuals in the top third of baseline plasma Lp(a) compared with those in the bottom third (4.8).
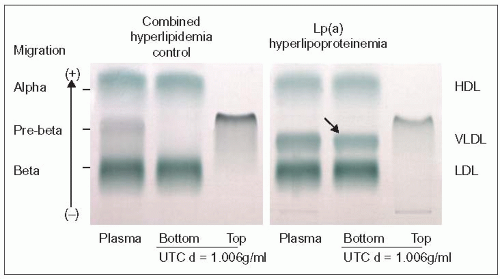 4.2 Demonstration of Lp(a) as a ‘sinking preβ-lipoprotein’. Using a combination of ultracentrifugation of plasma at density d = 1.006 g/ml and agarose gel electrophoresis as described in 2.17, one can demonstrate that Lp(a) shows preβ migration, but sinks to the bottom of the ultracentrifugation (UTC) tube (right panel, arrow), hence its previous name of ‘sinking preb lipoprotein’. The lipoprotein profile was as follows: cholesterol 6.93 mmol/l, low-density lipoprotein-cholesterol (LDL-C) 5.17 mmol/l, triglycerides 0.89 mmol/l, and HDL-C 1.11 mmol/l (268 mg/dl, 200 mg/dl, 79 mg/dl and 43 mg/dl, respectively); the Lp(a) was 100 mg/dl. The left panel shows a control with combined hyperlipidemia (high LDL and very-low-density lipoproteins [VLDL]). The preb lipoprotein seen in total plasma floats in the top fraction and is not seen in the bottom fraction. This technique was popular before commercial kits became available for the measurement of Lp(a). It is important to know because when analysing whole plasma by lipoprotein electrophoresis the band of Lp(a) may be mistaken for an excess of VLDL. |
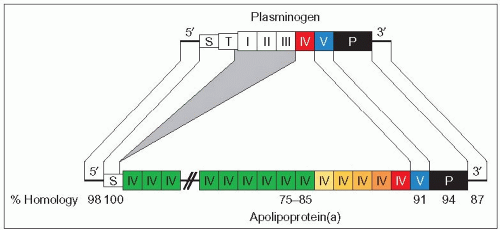 4.3 Homology between plasminogen and apo(a) genes. Discovery of the homology between the apo(a) gene and the plasminogen gene was a breakthrough that provided new insight into the role of Lp(a) in athero-thrombogenesis. Both plasminogen (PLG) and apo(a) (LPA) genes are member of the same gene superfamily and both are localized on chromosome 6. Both genes have nucleotide sequences coding for different kringle domains, a signal sequence (S) allowing secretion and a protease domain (black rectangle). Whereas plasminogen is a plasma serine protease zymogen, apo(a) has an inactive protease domain. Both genes share a sequence coding for a kringle (K) V (blue) and a kringle IV (in red in both genes to illustrate the marked similarities between K IV of PLG and K IV type 10 of apo(a) which is closest to K V). LPA does not share the ‘tail region’ (T) and kringles I, II and III of PLG. The other major difference is in the diversity of the kringle domain sequences for PLG and the numerous (and variable) repetitions of the sequences coding for a kringle IV domain in LPA. Also, kringles of apo(a) are glycosylated but kringles of plasminogen are not. Connecting lines indicate regions of homology with shading in grey to represent domains that are lacking in apo(a). The percentage of identity of plasminogen and apo(a) cDNA sequences for the following domains is shown at the bottom: 98% for the 5′-untranslated region, 100% for the signal sequence, 75-85% for kringle IV, 91% for kringle V, 94% for the protease sequence, and 87% for the 3′-untranslated region. This high degree of homology has raised the possibility that the apo(a) gene originated via duplication and remodelling of the plasminogen gene. It is thought that they evolved from a common precursor about 40 million years ago when Old and New World monkeys diverged. Redrawn from McLean JW et al. (1987). cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature, 330: 132-137. |
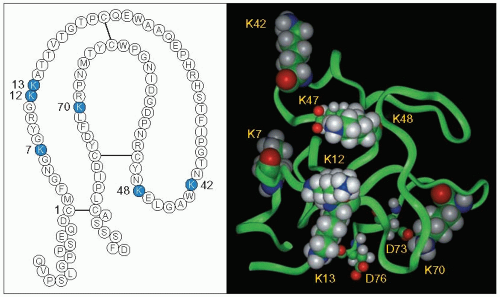 4.4 Amino acid sequence and spatial model of kringle V. Kringle V immediately proximal to the protease domain is important in apo(a) because it is the site for covalent binding of oxidized phospholipids and is pro-inflammatory, both consistent with an atherogenic potential. The amino acid sequence and the loop structure of the kringle domain stabilized by intrachain disulphide bonds are given in the left panel. This three-loop structure resembles the Danish pastry bearing this name. The numbering begins with Cys (C) in position 1. The six Lys (K) in positions 7, 12, 13, 42, 48, and 70 are shown as blue circles. The amino acid residues preceding Cys-1 and following Cys-80 are portions of the linker regions joining K IV10 to Cys-1 and Cys-80 to the protease domain. These kringle structures are also present in plasminogen, prothrombin, tissue plasminogen activator, urokinase, coagulation factors VII, IX, X, XII and protein C. These peptides are members of a protein superfamily acting as regulatory proteases in the fibrinolytic and coagulation systems. Kringles help binding of these peptides to other macromolecules or receptors. In the right panel, the spatial model of K V derived from the crystal structure of human apo(a) K IV10 is given. The six Lys are shown by space-filled atoms. Lys-48 and Lys-70 form salt bridges with the carboxyl side chains of Glu-47 (E) and Asp-73 (D), respectively (shown in smaller space-filled atoms). Lys-13 makes hydrogen bonds with the main chain carbonyl of Asp-76. Lys-7 being located in the inner surface groove is excluded from interactions. Lys-12 and Lys-42 protrude from the kringle surface and appear to be free of constraints and may be the likely candidates for covalent linkage to oxidized phospholipids. White = hydrogen; green = carbon; blue = nitrogen; red = oxygen. Reproduced from: Edelstein C et al. (2003). Lysine-phosphatidylcholine adducts in kringle V impart unique immunological and potential pro-inflammatory properties to human apolipoprotein(a). J Biol Chem, 278: 52841-52847. |
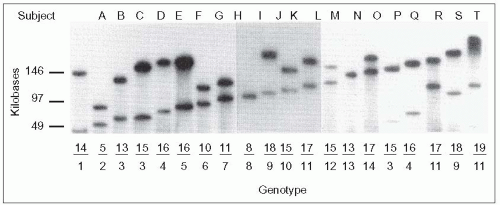 4.5. Multiple alleles and high heterozygosity of the apo(a) gene. Genomic DNA from 102 unrelated Caucasians was obtained from blood mononuclear cells, digested with the restriction enzyme KpnI and separated according to size by pulse-field gel electrophoresis, transferred to a nylon membrane, and the restriction fragments revealed by hybridizing with a 32P-labelled apo(a) kringle IV specific probe. This study, carried out by Lackner and colleagues at Hobbs’ laboratory at the University of Texas Southwestern Medical School, revealed the high degree of heterozygosity of the apo(a) gene. In these 102 individuals, 19 different alleles were identified and 94% of these individuals had two different alleles. Lackner also demonstrated that the size of the apo(a) gene correlated directly with the size of the apo(a) protein and inversely with concentration of Lp(a) in plasma. Interestingly, an allele can be associated with similar levels of Lp(a) among members of a given family, but the same allele in another family may be associated with markedly different levels that also remain consistent among family members. This suggests that levels may be determined by interaction with another factor (i.e. a ‘second hit’). Although more insight might be obtained regarding the inheritance of Lp(a) in families using this technique and perhaps refine cardiovascular risk predictability as some studies have indicated, it has not received much interest from the standpoint of clinical practice. This is owing to the fact that the technique is labour-intensive, time-consuming, expensive, needs to be done in dedicated laboratories and has not been simplified for the study of large numbers of subjects. Measurement of the Lp(a) mass is the only approach used at present. Lp(a) may be quantified by a variety of techniques using either antibodies to the kringle IV type 2 domain (unfortunately influenced by size polymorphism), a latex-enhanced immunonephelometric assay using monoclonal anti-Lp(a) (simple, automated), or isolation of intact Lp(a) particles by lectin affinity and measurement of its cholesterol content. This topic was critically reviewed by Lippi and Guidi ([2003]. Critical Rev Cin Lab Sci, 40:1), and these authors raised important practical points: the need to assess confounding factors to avoid undue delays before measurements after sampling; recognition of temperature sensitivity (Lp(a) is sensitive to prolonged storage at room temperature or at 4 °C [oxidation, degradation]; best storage is at -70°C; repeated freezing and thawing is deleterious); awareness that the method of calibration is difficult because of the 1000-fold variation in plasma levels; and use of a definitive standard is necessary. This figure is reproduced from Lackner C et al. (1991). Molecular basis of apolipoprotein(a) isoform size heterogeneity as revealed by pulsed-field gel electrophoresis. J Clin Invest, 87: 2153-2161. |
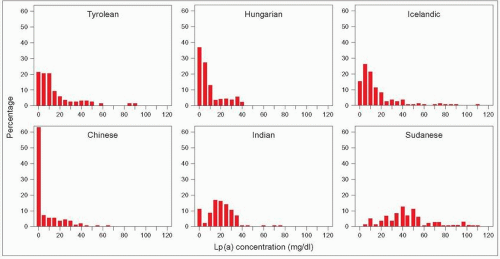 4.6 Frequency distribution of lipoprotein(a) (Lp(a)) plasma levels vary widely across populations. The frequency distribution of Lp(a) plasma concentrations was measured in seven different ethnic groups by Sandholzer C et al. in 1991 (six are shown here). The distribution is skewed to the right in most populations warranting log transformation for statistical analysis. However, this distribution varies widely across populations of different ethnic origin; this is particularly striking here when comparing the Chinese and black Sudanese cohorts. In the latter the distribution is nearly Gaussian. The authors also carried out apo(a) phenotyping in these populations. They observed that: there is considerable heterogeneity of Lp(a) polymorphism among populations; differences in apo(a) allele frequencies alone did not explain differences in Lp(a) levels among populations; and in some populations, such as the Sudanese blacks, Lp(a) concentrations are determined by factors that are different from the apo(a) size polymorphism. Redrawn from Sandholzer C et al. (1991). Effects of the apolipoprotein(a) size polymorphism on the lipoprotein(a) concentration in 7 ethnic groups. Hum Genet, 86: 607-614. |
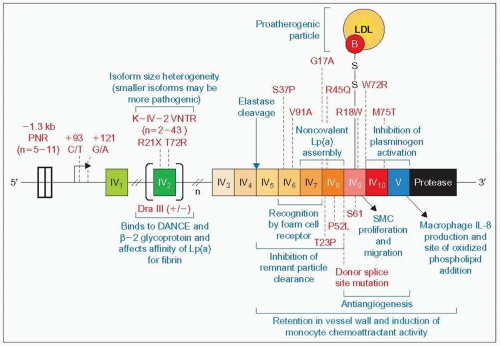 4.7 — Figure caption opposite. 4.7 (Figure bottom of previous page) Apo(a) gene mutations and structure-function relationships. The apo(a) gene size varies between 32 kb and 189 kb and differs by increments of 5.6 kb corresponding to one kringle unit. Each kringle is coded by two exons, and because of the variable number of kringle repeats the total number of exons also varies. This composite and hybrid diagram, which refers to the gene as well as to the protein, is used to display the polymorphic sites and mutations of the apo(a) gene (or protein) in red and the structure-function relationships in blue. The disulphide bond that links kringle (K) IV type 5 of apo(a) to apoB of LDL is indicated as part of the review of the structure-function relationships. On the apo(a) gene in the middle of the diagram the location of the regions coding for the ten different types of kringles from 1 (IV1) to 10 (IV10) are depicted. The polymorphisms, mutations and sources of variation are given (red text) from the 3′ to the 5′ end of the gene. PNR denotes the penta-nucleotide repeat polymorphism in the promoter, the number ranging from 5 to 11. K IV2 VNTR denotes the variable number of tandem K IV type 2 repeat polymorphisms responsible for size variation of apo(a) (n = 2-43). DraIII refers to a polymorphic site detected using the restriction enzyme DraIII present in a subset of K IV type 2 kringle sequences. It occurs with higher frequency in Chinese and is invariably associated with low plasma levels of Lp(a) in Caucasians. The substitution of C by T at position +93 leads to negative regulation in expression of the gene, while a change of G to A at position +121 leads to positive regulation. The T72R mutation of K IV2 abolishes the lysine binding capabilities of apo(a) and should therefore produce a more benign Lp(a) with less athero-thrombogenic potential. The R21× (Arg21→Ter) in exon 1 of K IV type 2 domain and the donor splice site mutation of the 6 kb intron separating the two exons of K IV type 8 repeat cause Lp(a) deficiency. These subjects appear healthy. There are sequence variants in K IV6 to K IV10 exons whose effects are neutral or undetermined except for a rare mutation W72R in K IV10 (Trp72→Arg) affecting the lysine binding of apo(a) which has been observed in some individuals from the black population in the USA. This part of the figure is redrawn and updated from Utermann G (1999). Genetic architecture and evolution of the lipoprotein(a) trait. Curr Opin Lipidol, 10: 133-141. Specific functions that have been ascribed to discrete functional units of Lp(a) are given in blue on this diagram. These functional domains have been identified on apo(a) using a combination of the expression of recombinant variants of apo(a) and elastase cleavage of apo(a) and Lp(a). Elastase cleaves the protein between K IV4 and K IV5 as shown. The atherogenic role is attributed in part to the: (1) isoform size heterogeneity that relates smaller size to higher Lp(a) levels and greater atherogenic potential, (2) presence of a site (K IV6 and K IV7) that recognizes a specific receptor on monocyte/macrophages favouring uptake of the pro-atherogenic Lp(a) with its LDL moiety to form foam cells, (3) interference of apo(a) with normal interaction of apoB with the LDL receptor, (4) ability of a region (K IV types 5, 6, 7 and 8) to delay clearance of circulating remnant lipoproteins (overexpression of this fragment in mice fed a high-fat diet enhances atherosclerosis), (5) K IV type 9 that stimulates smooth muscle cell (SMC) migration and proliferation, (6) induction of cytoskeletal rearrangements in the endothelial cells increasing their permeability and resulting in a dysfunctional endothelium, (7) presence of K V that stimulates interleukin (IL)-8 production by macrophages, and allows addition of oxidized phospholipids (the pro-atherogenic oxidized phospholipids are preferentially associated with Lp(a) compared with free LDL) (see 4.4, K12 and K42). All of the above and the propensity of apo(a) to adhere to arterial wall components such as fibrin, fibrinogen, fibronectin and glycosaminoglycans favour retention of Lp(a) in the arterial wall. The same applies to binding of apo(a) and Lp(a) via the K IV2 repeats to β2-glycoprotein-1 (also called apoH) and to the extracellular matrix protein DANCE (developmental arteries and neural crest epidermal growth factor [EGF]-like). Regarding the antifibrinolytic and thrombogenic role, the following mechanisms apply: (1) apo(a) interference with the efficient activation of plasminogen to plasmin attributed to the combined effect of K IV10 and K V binding to the complex plasminogen, tissue-type plasminogen activator (t-PA) and fibrin, (2) competition of apo(a) for binding of plasminogen to fibrin and fibrinogen via K V and the protease domain, (3) enhancement of plasminogen activator inhibitor (PAI)-1 activity. In addition, the fragment containing K IV9, K IV10 and K V inhibits basic fibroblast growth factor-stimulated venous endothelial cell proliferation and migration and represses neovascularization in chick embryos and tumour growth in nude mice. These observations suggest antiangiogenic and anti-carcinogenic effects. It has also been speculated that Lp(a) transports cholesterol to sites of injury and wound healing, but as an untoward side effect it may also trigger deposition of cholesterol in growing atherosclerotic plaques. This part of the diagram has been modified slightly from Koschinsky ML, Marcovina S (2004). Structure-function relationships in apolipoprotein(a): insights into lipoprotein(a) assembly and pathogenicity. Curr Opin Lipidol, 15: 167-174, and Koschinsky ML (2005). Lipoprotein(a) and atherosclerosis: new perspectives on the mechanism of action of an enigmatic lipoprotein. Curr Atheroscler Rep, 7: 389-395. |
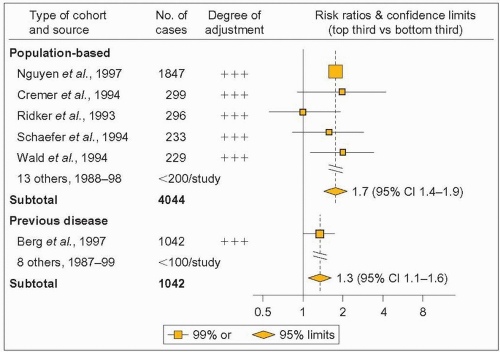 4.8 Meta-analysis of prospective studies on coronary artery disease (CAD) risk impact of lipoprotein(a) (Lp(a)). For many years it was controversial whether Lp(a) is an independent CAD risk factor because both negative and positive studies had been published. This large meta-analysis, along with other recent studies, has resolved the issue. It took into account geographical location of study, size, type of cohort (population-based or selected because of previous disease), mean age, follow-up duration, blood storage temperature and duration. It included 5436 deaths from coronary heart disease or non-fatal myocardial infarctions during a weighted mean follow-up of 10 years in 27 eligible studies. The risk ratios compare top and bottom thirds of baseline measurements. Orangeyellow squares indicate the risk ratio in each study, with the size of the squares proportional to number of cases and horizontal lines representing 99% confidence intervals (CIs). The combined risk ratios and their 95% CI are indicated by orange-yellow diamonds. Degree of adjustment for possible confounders is denoted as follows: +, adjustment for age and sex only; ++, adjustment for the preceding plus smoking; and +++, adjustment for the preceding plus some other classical vascular risk factors. For sake of simplicity, population-based studies including fewer than 200 cases or previous disease studies including fewer than 100 cases are not shown on this partial reproduction of the figure published by Danesh J et al. In the prospective studies not shown (n = 11-191) all but one had a +++ level of adjustment. Comparison of individuals in the top third of baseline plasma Lp(a) measurements with those in the bottom third in each study yielded a combined risk ratio of 1.6 (95% CI 1.4-1.8, 2P < 0.00001). Reproduced with permission from Danesh et al. (2000). Lipoprotein(a) and coronary heart disease – meta-analysis of prospective studies. Circulation, 102: 1082-1085. |
In the large Prospective Epidemiological Study of Myocardial Infarction (PRIME), subjects with levels of Lp(a) in the highest quartile had more than 1.5 times the risk than subjects in the lowest quartile. Moreover, the risk was a function of LDL-C levels as observed previously. A Lp(a) concentration above 33 mg/dl combined with a LDLC >163 mg/dl (>4.22 mmol/l) was associated with a 4.5-fold increase in CAD risk compared with a relative risk of 0.82 when combined with an LDL-C <121 mg/dl (<3.13 mmol/l) (4.9). Some studies of familial hypercholesterolemia (FH) with or without CAD have shown a higher frequency of high Lp(a) levels in the presence of CAD (4.10). The association between Lp(a) and cardiovascular disease and stroke in particular may be stronger in men than in women.
The mechanism whereby Lp(a) may promote cardiovascular disease (CVD) has been ascribed to both its LDL-related structure and its plasminogen-like antifibrinolytic function. Lp(a) (and free apo(a)) is found in the atherosclerotic arterial wall, and kringle IV-type 10 has a dominant role in the binding of apo(a) to fibrin or fibrinogen and components of the extracellular matrix (4.7). Lp(a) is also readily oxidizable in vitro similar to LDL (4.11) and is taken up by monocyte-macrophages, resulting in a potentially atherogenic particle (4.12). The expression of tissue plasminogen activator (t-PA) and inhibitor-1 (PAI-1) in the vascular wall is influenced by the presence and concentration of Lp(a), which is retained more avidly than LDL. Small apo(a) size polymorphism has also been found to be associated with CVD in men. The kringle V of Lp(a) (4.4 and 4.7) covalently binds oxidized phospholipids (especially phosphatidylcholine), which may accumulate into the vessel wall and promote atherosclerosis. Kringle V also stimulates IL-8 production by human macrophages in culture, a pro-inflammatory effect. An association of Lp(a) with coronary calcifications in human atherosclerosis specimens has been reported, consistent with observations in the WHHL transgenic rabbit expressing human apo(a) (4.13). Although three studies failed to find an association between coronary calcification and Lp(a), one of them, the Dallas Heart Study, showed that there was a higher prevalence of coronary artery calcium (CAC) measured by electron beam computed tomography (EBCT) among Caucasians with high plasma levels of Lp(a) plus elevated plasma levels of LDL-C (men) or reduced levels of HDL-C (men and women). Finally, in men with CAD and high LDL-C in the Familial Atherosclerosis Treatment Study (FATS), Lp(a) concentration was a major correlate of baseline disease severity, atherosclerosis progression, and event rate over 2.5 years. Moreover, with substantial LDL-C reductions, persistent elevations of Lp(a) were no longer atherogenic or clinically threatening.
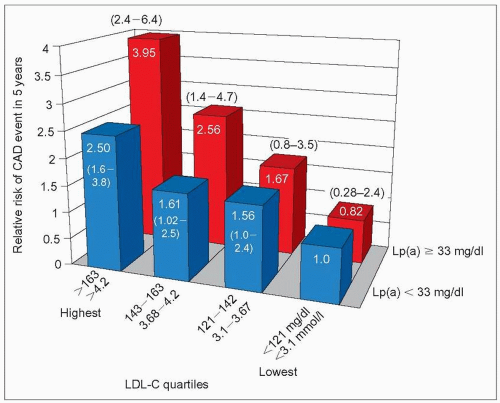 4.9 Lipoprotein(a) (Lp(a)) enhances coronary artery disease (CAD) risk associated with high low-density lipoprotein-cholesterol (LDL-C). Several studies have indicated an interaction between Lp(a) and LDL-C, a notion of prime importance in clinical practice. The PRIME study (Prospective Epidemiological Study of Myocardial Infarction) provided a clear demonstration of this interaction as illustrated here. PRIME included 9133 French and Northern Irish men aged 50-59 at entry, without a history of CAD and not on lipid-lowering drugs. During 5 years of follow-up, 288 subjects experienced at least one CAD event (myocardial infarction, coronary death, angina pectoris). Logistic regression analysis was used to evaluate Lp(a) levels as a predictor of CAD. Lp(a) was a highly significant predictor of CAD events in the entire cohort (P < 0.0006) with a relative risk between top and lower quartile of Lp(a) of 1.56 (95% CI 1.10-2.21). As shown on this graph derived from data reported by Luc G et al. the relative risk (RR) of a CAD event associated with a Lp(a) level of <33 mg/l increased gradually from the referent value of 1.0 to 2.50 (95% CI 1.6-3.8) from the lowest (<121 mg/dl; <3.1 mmol/l) to the highest (>163 mg/dl; >4.2 mmol/l) quartile of low-density lipoprotein-cholesterol (LDL-C). In contrast, the RR of a CAD event associated with a Lp(a) level ≥33 mg/dl increased more steeply from 0.82 (95% CI 0.28 to 2.4) to 3.95 (95% CI 2.4-6.4) from the lowest to the highest quartile of LDL-C. Therefore, high levels of Lp(a) increase the risk of CAD associated with high LDL-C (upper half of the distribution), conversely high levels of Lp(a) in the absence of elevated LDL-C (lower half of the distribution) do not add to the risk already associated with the LDL-C level. Bar graph drawn from data in Luc G et al. (2002). Lipoprotein(a) as a predictor of coronary heart disease: the PRIME Study. Atherosclerosis, 163: 377-384. |
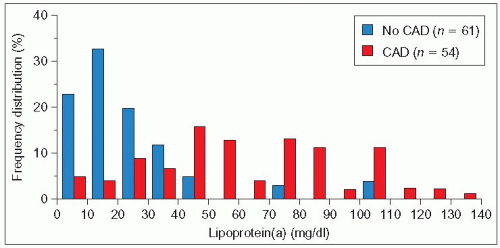 4.10 Higher frequency of elevated lipoprotein(a) (Lp(a)) levels in familial hypercholesterolemia (FH) patients with coronary artery disease (CAD) than in patients without CAD. The frequency distribution of serum Lp(a) concentration in heterozygous FH subjects from a UK lipid clinic without CAD (blue columns) resembles that observed in normal Caucasians (see 4.6) with only a few individuals having very high levels. In contrast, the frequency distribution in FH subjects with CAD (red columns) is clearly displaced towards higher Lp(a) concentrations. This is reflected in the median Lp(a) levels being 18 mg/dl in the absence of CAD and 57 mg/dl in its presence. This finding has been confirmed in some studies but not in others. This may reflect the differences in ethnicity, age, type of LDLR mutation or variation in other associated modulating factors. In practice, the family history and severity of the disease in a proband should guide the need to measure Lp(a) in other family members. Redrawn from Seed M et al. (1990). N Engl J Med, 322: 1494-1499. |
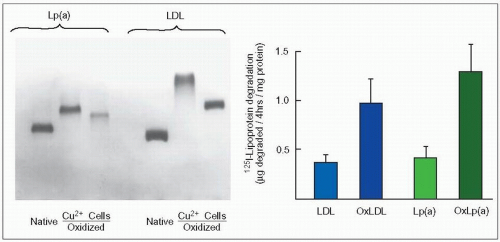 4.11 In vitro oxidation and macrophage uptake of native or oxidized lipoprotein(a) (Lp(a)) and low-density lipoproteins (LDL). The left panel illustrates that native LDL and Lp(a) isolated from the same patient with familial combined hyperlipidemia (FCH) and Lp(a) hyperlipoproteinemia can be oxidized in vitro when exposed to 10 μM CuCl2 (Cu++) or to human mononuclear cells (Cells), as shown by a faster electrophoretic mobility on agarose gel electrophoresis. This effect, which is completely abolished by the antioxidant probucol, modifies the structure of Lp(a) and enhances its atherogenic properties. Indeed, oxidized Lp(a) (OxLp(a)) as well as oxidized LDL (OxLDL) are taken up readily and degraded by human monocyte macrophages (right panel) to induce accumulation of cholesteryl esters and form foam cells. They have also been isolated from human atheromatous lesions (see 4.12). When the apo(a) was clipped off from LDL with dithiothreitol the remaining particle was degraded at a lower rate than the whole OxLp(a) indicating a preferential uptake and degradation. Work in the laboratory of Scanu at the University of Chicago has demonstrated that the size of the LDL moiety of Lp(a) may vary as the size of native LDL varies. It is likely that in FCH Lp(a) particles are smaller and denser as LDL are compared to those of a patient with familial hypercholesterolemia. These figures from work carried out in the authors’ laboratory are redrawn from Naruszewicz M et al. (1992). Oxidative modification of lipoprotein(a) and the effect of beta-carotene. Metabolism, 41: 1215-1224. |
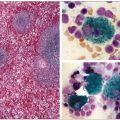
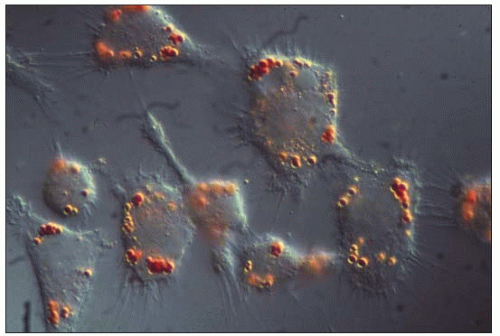 4.12 Macrophage uptake of lipoproteins isolated from human atheroma. In the early 1990s, Hoff at the Cleveland Clinic Research Institute isolated lesion low-density lipoproteins (LDL) and lipoprotein(a) (Lp(a)) from human atherosclerotic plaques obtained at autopsy. They were characterized and their properties evaluated. Lp(a) was present in a form resembling intact but oxidized Lp(a), larger particles representing Lp(a) complexed to itself or to other plaque components and a slightly smaller particle probably representing partially degraded Lp(a). He had similarly isolated and studied lesion LDL. The figure is an interference contrast photomicrograph of mouse peritoneal macrophages in culture exposed to lesion-derived LDL (100 μg/ml) and incubated 48 hours at 37°C and stained for lipids with Oil red O. Oxidized Lp(a) is similarly taken up by macrophages. The authors are grateful to Henry Hoff for having generously provided this unpublished picture. For further information, see Hoff HF, O’Neil J. (1991). J Arterioscler Thromb, 11: 1209-1222; Hoff HF et al. (1993). J Lipid Res, 34: 789-798; and Hoff HF et al. (1994). Chem Phys Lipid, 67-68: 271-280. |
The current Canadian clinical guidelines (but not all guidelines) include measurement of Lp(a) to quantify the CVD risk. A concentration of 30 mg/dl or more is considered high and an indicator of risk. The overall relationship of Lp(a) with cardiovascular disease and the quantitative contribution of elevated Lp(a) to CAD risk in the presence of high LDL-C are sufficiently well established (see 4.8–4.10) to warrant this attitude. A link between thromboembolic events and Lp(a) has been clearly demonstrated in several conditions; however, there are some caveats. Accurate standardized methods of measurements are not widely available in clinical chemistry laboratories and assays may be affected by apo(a) size heterogeneity (see 4.5). Several conditions may spuriously increase plasma Lp(a) levels, a confounding factor (see above). Serum Lp(a) is relatively resistant to lowering by diet, exercise, weight loss or statins. There is no evidence from clinical trials as yet to support a benefit from lowering Lp(a) levels; such a trial needs to be done but is difficult because nicotinic acid, the most effective agent, has multiple beneficial lipoprotein effects.
The diagnosis of ‘familial Lp(a) hyperlipoproteinemia’ is made difficult because of the variation in the apo(a) gene, its high heterozygosity and the dependence of the CVD risk on the size polymorphism of apo(a). Also, it does not have a specific clinical profile. Therefore, measurement of Lp(a) is particularly important in people with a moderate CVD risk, a strong family history of premature cardiovascular disease, and elevated LDL levels (especially if associated with low HDL-C), in those who have an unexpectedly severe atherosclerotic phenotype and in subjects from a family in which high Lp(a) segregates with manifestations of atherosclerosis. In these cases an elevated Lp(a) concentration should entail a more vigorous treatment of dyslipidemia and concomitant risk factors. Treatment with nicotinic acid (that may induce Lp(a) lowering up to 40%) and its slow-release derivatives, LDL apheresis, neomycin, and estrogen therapy in postmenopausal women can effectively decrease Lp(a) plasma concentration.
Familial phytosterolemia (ABCG5-ABCG8 defects)
Familial phytosterolemia (familial sitosterolemia, familial β-sitosterolemia, familial ATP-binding cassette family G type 5 and/or type 8 defect, sterolin deficiency), is an autosomal recessive disease characterized by elevated plasma plant sterols and relatively normal plasma cholesterol concentrations, xanthomatosis and premature CAD. It has raised much interest because study of this disease led to new discoveries that allowed a better understanding of cholesterol transport mechanisms in the liver and the intestine. The term phytosterolemia is preferred to sitosterolemia because all plant sterols are affected, not just sitosterol.
Healthy subjects have very low plasma levels of plant sterols (phytosterols), which are mostly derived from vegetable oils, seeds and nuts, mainly because they are poorly absorbed. Plant sterols also interfere with cholesterol absorption because of their ability to displace cholesterol from intestinal micelles. This led, before the era of fibrates and statins, to the use of a plant sterol emulsion as a cholesterol-lowering agent. The content of phytosterol in a normal diet is in the order of 150-450 mg/day. However, it takes 2 gm/day of this supplement to achieve a mere 10% cholesterol reduction on average. The need for multiple doses and flatulence were the trade-offs that caused its demise. The structural formula of plant sterols is shown in 4.14. The major plant sterols are sitosterol (because there are no a forms of plant sterols, this is no longer called β-sitosterol), campesterol and stigmasterol. They follow the same pathway of absorption and enterohepatic recirculation as cholesterol (4.15). They are taken up by micelles, enter the enterocytes via a specific transporter, Niemann-Pick C-1 like-1 protein (NPC1L1), and the bulk is rapidly returned to the intestinal lumen via the heterodimeric transporters ABCG5 (ATP-binding cassette family G type 5) and ABCG8 (also called sterolin-1 and sterolin-2). The small amount not re-excreted is esterified and transferred into chylomicrons and passes into the lymph to reach the circulation and the liver to be unesterified and re-excreted into the bile via liver ABCG5-ABCG8 transporter. The normal plasma level of phytosterol averages 0.7 mg/dl and represents less than 1% of the total sterol content of plasma lipoproteins. Excessive absorption of plant sterols may disrupt cholesterol homeostasis. Mice lacking the ABCG5 and ABCG8 half transporters have a 30-fold increase in plasma levels of plant sterols, and low biliary cholesterol concentration. Their plasma and liver cholesterol concentrations are reduced by 50% but rapidly increase with cholesterol feeding. They also display a massive reduction in the adrenal content of cholesterol. The human form of ABCG5-ABCG8 deficiency is familial phytosterolemia.
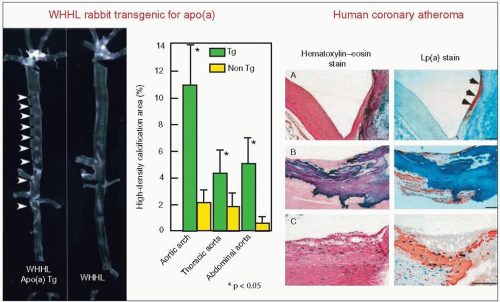 4.13 Lipoprotein(a) (Lp(a)) associates with calcifications in atherosclerotic lesions. The left panels provide a radiographic demonstration of multiple aortic calcification sites in the low-density lipoprotein (LDL) receptor-deficient Watanabe heritable hyperlipidemic rabbit (WHHL) transfected with the human apo(a) transgene (apo(a) Tg) compared with the non-transgenic (non Tg) WHHL. In this model, apo(a) markedly enhanced the spontaneous development of atherosclerotic lesions that resemble complex lesions observed in advanced human disease. The arrowheads point to the calcifications. The high-density areas (calcification) were quantitated on X-ray films by a computerized image analysis system and plotted in the bar graph. Values are expressed as a per cent of each segment of the aorta (P < 0.05 vs. the non Tg WHHL rabbits). They were significantly higher in the apo(a) transgenic WHHL in the three aortic segments studied. Immunostaining with antibodies against apo(a) and apoB showed that apo(a) was frequently deposited around the calcified areas and co-localized with apoB. The authors also found that human Lp(a)-treated rabbit aortic smooth muscle cells (SMC) showed increased alkaline phosphatase activity and enhanced calcium accumulation. In addition, Lp(a) tended to stimulate SMC toward osteoblastic differentiation by inducing Osf2 (osteoblast specific factor-2) mRNA expression. The right panel demonstrates that Lp(a) is intimately associated with areas of calcification in human coronary arteries as observed in the rabbit model. (A) Lp(a) deposition (orange) along the surface of calcified area on the right side (indicated with arrowheads), (B) Lp(a) around ossification, and (C) in this lesion, calcium deposition is sparsely distributed beneath the foam cells and Lp(a) deposition is intermingled with SMC. Scale bars represent 200 mm. Reproduced with permission from Sun H et al. (2002). Lipoprotein(a) enhances advanced atherosclerosis and vascular calcification in WHHL transgenic rabbits expressing human apolipoprotein(a). J Biol Chem, 277: 47486-47492. |
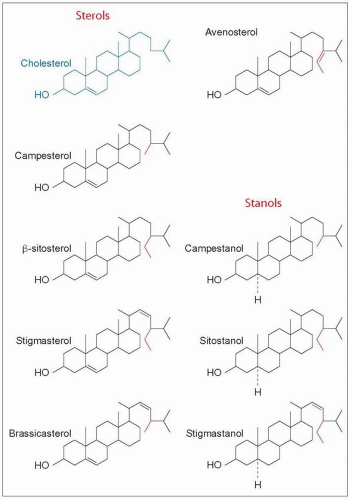 4.14 Comparison of the structure of plant sterols and stanols with cholesterol. The chemical structure of plant sterols is similar to that of cholesterol. Campesterol and β-sitosterol, the 24-methyl and the 24-ethyl analogues of cholesterol respectively, are the most abundant plant sterols. They occur at low concentrations in human plasma and they represent 95% of dietary phytosterols. Dehydrogenation of the carbon 22-23 bond of sitosterol leads to stigmasterol which is another common phytosterol. The others, brassicasterol, avenasterol, ergosterol, and stigmasterol, and also shellfish sterols, have been identified in plasma in minute amounts. Plant sterols are abundant in the diet and found in considerable amounts in seeds, nuts, fruits and vegetable oils. Daily intake ranges from about 180 mg in the USA to 480 mg in Japan. Saturation of the Δ5 double bond of the three major plant sterols leads to the formation of 5a-derivatives, sitostanol, campestanol and stigmastanol. Although present in the diet in small amounts they are almost totally unabsorbable. As they may effectively compete with cholesterol in the formation of mixed micelles and interfere with cholesterol absorption they have raised interest as cholesterol-lowering agents, and have been introduced in ‘functional foods’ such as sitostanol margarines. |
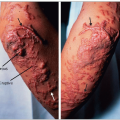
Familial phytosterolemia (OMIM No. 210250) is a rare autosomal recessive condition with a frequency estimated at 1:1 500 000, which shares the clinical features of familial hypercholesterolemia (FH). Tuberous, planar and tendinous xanthomatosis (4.16 and 4.17), developing in childhood, may be moderate or severe enough to determine a clinical phenotype that is indistinguishable from that of homozygous FH. A generalized eruptive xanthomatosis has even been reported in a 6-year-old child. Arthralgia and arthritis (knees and ankle joints), hypersplenism, thrombocytopenia and hemolysis may be present. Plasma cholesterol concentrations may be normal or moderately elevated, but in some children may reach levels found in homozygous FH (>13 mmol/l; >500 mg/dl) whereas triglycerides are normal. Elevated apoB is another feature of familial phytosterolemia. Premature atherosclerosis is typical and death from CAD has been reported as early as age 13 (4.18). The disease is characterized primarily by an increased intestinal absorption (15-16% instead of <5%) and a reduced biliary secretion of plant sterols. This results in plasma levels of sitosterol and campesterol approximately 50-fold higher than normal (14-65 mg/dl vs. 0.3-1.0 mg/dl for β-sitosterol). Plasma levels of other plant sterols such as stigmasterol, campesterol, sitostanol and campestanol (4.14) are also elevated. A high vegetable oil diet may worsen the disease. The plant sterols are carried by LDL and HDL particles and accumulate in tissues but not in the brain (although spinal intradural xanthomatosis has been reported in this condition). The etiology of this disease, which was first reported in 1974 by Bhattacharyya and Connor and by Kwiterovitch and colleagues, was unknown until recently when Hobbs and colleagues, at the University of Texas Southwestern Medical Center in Dallas, discovered that it segregated with mutations of the adenosine triphosphate (ATP) binding cassette transporters G5 and G8 (ABCG5, ABCG8) genes (4.19). Each gene codes for a ‘half transporter’ ABCG5 (sterolin-1) and ABCG8 (sterolin-2) that exert their function together in the heterodimeric form (4.20) to mediate efflux of dietary sterol in the intestine and protect humans against overaccumulation of sterols.
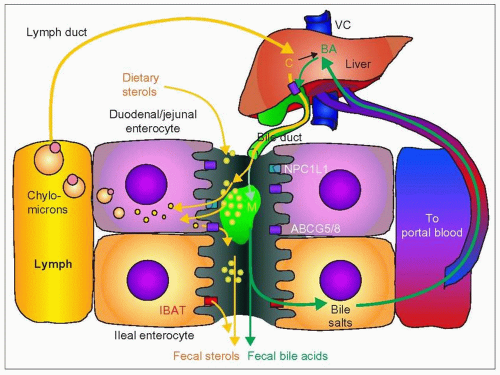 4.15 Sterol absorption and the two enterohepatic recirculations. In this diagram, the yellow path is that of cholesterol and the green path that of bile salts; they each have an enterohepatic recirculation that is key to understanding their respective roles in cholesterol homeostasis of the body. Intestinal cells are in contact with both lymphatics and blood capillaries. The duodenal and jejunal enterocytes (pink) handle lipids and bile acid differently from the ileal enterocytes (orange). Dietary cholesterol (∽400 mg/day) and other sterols (∽200 mg/day) enter the intestine and mix with free sterols and bile salts coming from the bile (1200 mg/day). The liver synthesizes about 400 mg/day of cholesterol. The unesterified sterols are taken up by enterocytes as part of mixed micelles (M) — formed in the lumen with bile salts, fatty acids, and phospholipids — by diffusion or by membrane transporter molecules such as Niemann-Pick C-1 like protein-1 (NPC1L1). The sterols are esterified in the enterocyte and packaged into chylomicrons for transfer into the lymph, a very active process after a fatty meal. This takes place mostly in the proximal small intestine. The chylomicrons reach the main circulation via the thoracic duct into the left subclavian vein thus eventually reaching the liver through the hepatic artery. Some of the sterols are also re-excreted into the intestine. Cholesterol absorption varies from 25% to 75%, fluctuates with time and is context dependent. Phytosterol absorption is usually less than 5%. Unabsorbed sterols and neutral sterols are excreted in the feces. About 800 mg of neutral sterols and acidic sterols (from bile salts) are excreted daily in the feces. Bile salts are synthesized from cholesterol in the liver. They enter the intestine in the bile, favouring absorption of cholesterol by their contribution to the formation of mixed micelles. In the intestine, 95% is removed mostly by active transport in the distal third of the small intestine, thanks to the highly effective ileal bile acid transporter (IBAT) also called apical sodium dependent bile acid transporter (ASBT). The remainder ends up in the feces as ‘acidic sterols’. Knockout of the IBAT gene (Slc10a2) in the mouse abolishes the enterohepatic recycling of bile acids. The reabsorbed bile salts are exported to the liver directly by the portal system to be reutilized in bile formation.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|