Reviews requests to research medical devices
Collects and acts on information related to safety and possible injury
Sets and enforces good manufacturing processes (GMP) and performance standards
Monitors compliance and surveillance programs
Provides technical support to small manufacturers of medical devices
manufacturers, institutions, and professionals and is aimed at reducing possible adverse effects. Specifically, medical device postmarket surveillance should
Establish a unique device identification system and promote its incorporation into electronic health information
Promote the development of national and international device registries for selected products
Modernize adverse event reporting and analysis
Develop and use new methods for evidence generation, synthesis, and appraisal
Provide timely, accurate, systematic, and prioritized assessments of the benefits and risks of medical devices throughout their marketed life using high-quality, standardized, structured, electronic health-related data
Identify potential safety signals in near real time from a variety of privacy-protected data sources
Facilitate the clearance and approval of new devices or new uses for existing devices
 EVIDENCE FOR PRACTICE
EVIDENCE FOR PRACTICEcommunity and the public, without facility and patient identification, so that clinicians nationwide may take necessary preventive actions.
may be made in an effort to solve a patient safety issue or as a cost-containment initiative. Such decisions include the following factors: cost, quality, efficacy, safety, and availability.
International Organization for Standardization (ISO)
http://www.iso.org/iso/home.html
Association for the Advancement of Medical Instrumentation (AAMI)
http://www.aami.org/
Emergency Care Research Institute (officially known as ECRI)
https://www.ecri.org
Advanced Medical Technology Association (AdvaMed) and formerly the Health Industry Manufacturers Association
http://advamed.org/
 EVIDENCE FOR PRACTICE
EVIDENCE FOR PRACTICEpharmacists, and biomedical engineers in an effort to manage the challenge and develop a system approach to safety. Stakeholders in IV medication-related events are listed in Table 12-2.
TABLE 12-1 COMMON BARRIERS TO SAFE MEDICATION ADMINISTRATION | ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 12-2 STAKEHOLDERS IN INTRAVENOUS MEDICATION-RELATED EVENTS | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
 PATIENT SAFETY
PATIENT SAFETYunderstand the requisite education necessary to fully benefit from and improve IV smart pump use and clinical integration. Failure to use IV smart pumps places the nurse and patient at increased risk (Harding, 2013).
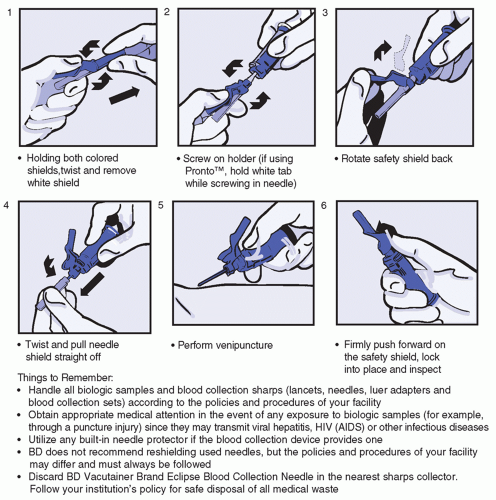 FIGURE 12-1 Vacutainer Eclipse blood collection needle, economical needle-based safety device for injection in a variety of sizes. (Adapted from BD Vacutainer Eclipse Blood Collection Needle Quick Reference Card. Courtesy of and © Becton, Dickinson and Company.) |
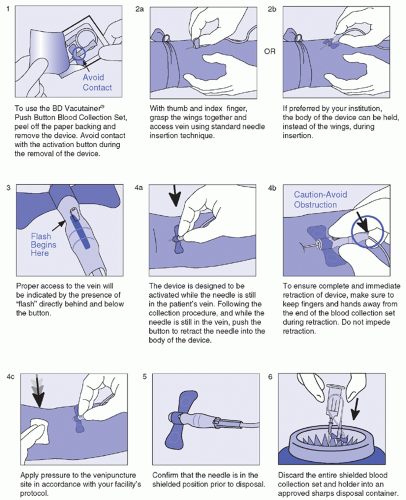 FIGURE 12-2 BD Vacutainer push-button blood collection/in-vein needle activation. (Adapted from BD Vacutainer Push Button Blood Collection Set InService Poster. Courtesy of and © Becton, Dickinson and Company.) |
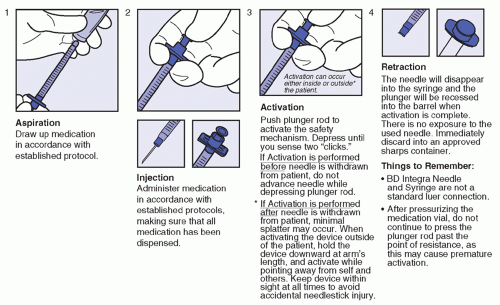 FIGURE 12-3 BD Integra 3-mL syringe, a low-waste space syringe with detachable retracting needle. (Adapted from BD Integra 3 mL Syringe Quick Reference Card. Courtesy of and © Becton, Dickinson and Company.) |
Fortunately, the human body readily adapts to changes in pressure, which enables clinicians to deliver IV therapy safely.
Intended for blood collection and short-term infusion
Facilitates the prevention of accidental needlestick injury
Ease-of-use reduces training time while improving activation compliance
Easily activated without patient discomfort
Ideal for use in high-risk environment
Latex-free
Flash visualization confirms venous access prior to collection of specimen
Blood flashback is seen clearly through the translucent body
One-handed safety activation also allows attention to the patient and venipuncture site
monitoring strip, should be attached to the container and hourly increments marked on the tape or strip, beginning with the time the infusion began.
would take approximately 1.5 hours to administer 1 L of 5% dextrose to a person weighing 70 kg or twice as long for 1 L of 10% dextrose. This maximum rate is faster than usual and is not customarily used except in an emergency.
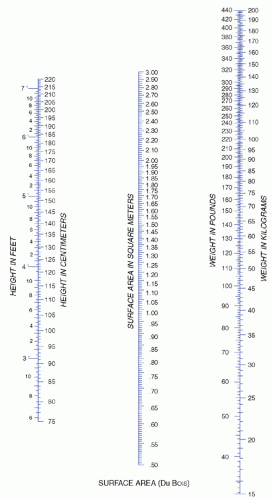 FIGURE 12-4 A nomogram. |
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


