Fig. 17.1
Evolution of inflammation in pancreatic ductal adenocarcinoma (PDAC). Progression through pre-invasive pan-IN stages to PDAC is associated with loss of cell mediated immunity, decreased numbers of dendritic cells and CD8+ T cells and increased tumour associated macrophages and immunosuppressive bone marrow derived cells (BMDCs) and T regulatory cells (Treg). PDAC that forms is stromally dense, and thick with extracellular matrix proteins and up-regulated signalling pathways
In early tumourigenesis, immune cells, including CD8+ T cells, detect danger signals from cancer cells and respond by eliminating these cells, leading to tumour immunogenicity. Dendritic cells are key regulators of tumour-specific immune responses in that they function as antigen presenting cells, activating CD8+ cytotoxic T lymphocyte (CTL) responses or stimulating CD4+ T cells through interaction with major histocompatibility complex (MHC) class II molecule-antigen complexes (Finn 2008). In pancreatic tumourigenesis, Hiraoka and colleagues reported that chemokine CXCL17 was responsible for infiltration of immature dendritic cells while ICAM-2 permitted killing of tumour cells by CD8+ cells. During evolution from precursor lesions to PDAC they found that the host immune response was tempered and tumours developed immune tolerance by downregulating CXCL17 and ICAM-2 (Hiraoka et al. 2011). Interestingly, Dillman and colleagues recently described the value of reintroducing dendritic cells loaded with antigen from autologous irradiated tumour cells in metastatic melanoma. Those patients who received this dendritic cell vaccine survived significantly longer than irradiated tumour cell vaccine alone (Dillman et al. 2011). Conversely, in PDAC, blockade of TLR4 signalling enhanced dendritic cell-mediated recruitment of T helper 2 (Th2) CD4+ cells, and this resulted in increased pancreatic inflammation and accelerated tumour progression (Ochi et al. 2012a). Thus depending on antigenic stimulus, dendritic cells can both promote and inhibit tumour progression. Promotion of anti-tumoural cell mediated responses, such as are generated through effective tumour immunosurveillance are being pursued in cancer. These strategies focus on engaging T cell responses in a field referred to as cancer immunology. The use of tumour antigen based vaccines and T cell adoptive transfer has provided some success in a small cohort of patients, however, the majority fail to respond clinically. More successful has been the use of T cell checkpoint inhibitors, to promote T cell activation. CTLA4 inhibitors such as ipilimumab and Programmed cell death 1 (PD1)/Programmed cell death ligand (PDL1) checkpoint inhibitors may be used to enhance CTL responses. Indeed the advances that these drugs have made in the field of metastatic melanoma have been striking. CTLA4 is a CTL surface protein and functions as a negative regulator of T cell function. CTLA4 interacts with APC membrane bound co-stimulatory molecules and functionally competes with the T-cell co-stimulatory molecule CD28. Thus ipilimumab binds and inhibits CTLA4 releasing a break to T-cell co-stimulation. Ipilimumab was the first agent to prolong overall survival in metastatic melanoma. CTLA4 is not the only improvement in the understanding of checkpoint biology. PD1 is an inhibitory receptor which following chronic T cell activation is induced via ligands including PDL1. Production of PDL1 by tumour associated macrophages or tumour cells themselves permits immune evasion by this mechanism (Sullivan et al. 2013), thus making it an exciting target for therapy. Trials in melanoma thus far have shown reliable tumour responses in at least 50 % of cases.
Commonly difficulties with these types of therapies arise when there are insufficient T lymphocytes infiltrating tumours to activate. Indeed, certain authors have suggested that high T cell infiltrate denotes good prognosis in these patients, while low infiltrate suggests failure of this strategy (Gajewski and Schumacher 2013). Two major mechanisms of immune resistance appear to exist in this context: failure of T cell trafficking; and immune suppression within the microenvironment. It may be hypothesised that removal of immunosuppressive cells such as bone marrow derived cells (BMDCs) and macrophages from the tumour microenvironment may allow activation of a more sustained T cell response.
17.3.3 Inflammatory Tumour Microenvironment and Therapeutic Opportunities
17.3.3.1 Functions of the Cellular Component of Tumoural Inflammation, Using Pancreatic Cancer as an Example
Once tumour immune surveillance is overcome, the composition of the immune infiltrate changes and a pro-tumourigenic leukocyte profile emerges. It is not yet fully understood how these different pro and anti-tumoural components of the immune system are engaged. One important question without resolution at present is the degree to which the plasticity of certain immune cells can be manipulated for therapeutic gain, and whether these properties will hold across a number of different tumour types or remain tissue specific. Thus, the majority of the following data draws in the main from studies in PDAC, seemingly the most dense and active of all solid tumour microenvironments, and a tumour in which excellent murine models of disease have been established to permit its study.
Signals in the pancreatic tumour microenvironment, including IL-10, can prevent dendritic cell activation, thus dampening the adaptive immune response (Koido et al. 2010). T cells can be both tumour suppressing and promoting depending upon their downstream target cells. CD4+ cells, particularly T regulatory cells and Th2 cells increase in number over the course of pancreatic cancer progression, while CTL cells decrease (Clark et al. 2007). This dynamic circumstance leads to a progressive accumulation of immunosuppressive cells that inhibit the anti-tumoural immune response. In tumours infiltrated by T regulatory cells both innate and adaptive immunity may well be suppressed, while BMDCs can also subdue anti-tumour activity (Gabrilovich and Nagaraj 2009).
In this milieu of tumour cell killing and escape, the immune system must clear damaged tissue. Neutrophils move into tissues under the control of chemokine signalling. Though short lived, neutrophils have the potential to induce significant tissue remodelling. Fridlender et al. demonstrated that following TGF-β blockade, neutrophils exhibited an anti-tumour phenotype termed N1 (Fridlender et al. 2009). Neutrophils are terminally differentiated cells, thus, they have often been overlooked with regards their role in cancer-related inflammation. However, neutrophils may also polarise to a pro-tumoural phenotype N2 in response to certain stimuli (Mantovani 2009), one of these stimuli is clearly TGF-β signalling. Chemokine receptor CXCR2 is expressed on neutrophils mediating chemotaxis of these cells to the tumour microenvironment (Jamieson et al. 2012).
Tissue macrophages evolve from blood monocytes, differentiating under the influence of signals received from the tissue microenvironment. Macrophages can be derived from existing populations initially established by the yolk sac during embryogenesis such as Kupffer cells in the liver. Macrophages following stimulation by bacterial lipopolysaccharides, and Th1 cytokines such as interferon gamma (IFNγ) adopt an M1 polarisation phenotype. M1 macrophages are broadly anti-microbial and anti-tumourigenic. Cytokine signals received from Th2 cells such as IL-13 and IL-4 drive an alternatively activated state, M2 macrophages, which largely carry out tissue remodelling purposes in health and following injury. These cells themselves express differing chemokine profiles. M1 macrophages produce pro-inflammatory chemokines including CXCL9 and CXCL10, in addition to IL-6 and TNFα among others following inflammatory stimuli, while M2 macrophages express non-inflammatory chemokines including CC chemokine ligand (CCL) 17, CCL18, CCL22, and CCL24 (Biswas et al. 2013). M2 macrophages fail to respond to inflammatory stimuli instead producing anti-inflammatory cytokines such as IL-10. It is possible for macrophages to exist on a spectrum between these two states, their functionality reliant on the pathophysiological stimulus.
Tumour-associated macrophages (TAMs) are present in early PDAC and persist throughout progression, representing an important component of the PDAC associated leukocyte infiltrate (Clark et al. 2007). Intriguingly, TAMs evolve to exhibit an M2 phenotype that is pro-tumourigenic in terms of promotion of growth and angiogenesis and suppression of adaptive immunity (Sica and Mantovani 2012). In cancer, this polarization of macrophages from a tumour-suppressive M1 phenotype may also be initiated by cytokine signals, including IL-10 and TGF-β, received from T regulatory and tumour cells (Sica and Mantovani 2012). This situation can be thought of as self-reinforcing, since production of pro-angiogenic molecules, including vascular endothelial growth factor (VEGF), attracts yet more macrophages to the tumour microenvironment, further enhancing angiogenesis.
Colony stimulating factor (CSF) 1 and its receptor CSF1R act as master regulators of macrophage differentiation from myeloid precursors. Via production of CSF1 and other chemokines including CCL2, tumours are able to chemo-attract macrophages to the tumour microenvironment (Hamilton and Achuthan 2013). CCL2 overexpression has been associated with progressive macrophage accumulation and progression of disease for some time now (Lin et al. 2001). CCL2 has also been identified as important in the recruitment of Ly6C+ macrophages from blood to sites of metastasis (Qian et al. 2011). The origin of TAMs is a matter of some debate, with the potential of both recruitment from blood and in situ proliferation possible. Indeed, much of the literature recently has pointed towards infiltration of BMDCs of a Ly6C+ subtype from the spleen to the tumour microenvironment resulting in macrophage differentiation in situ (Movahedi et al. 2010). The plasticity of TAMs makes them an ideal therapeutic target for manipulation in order to generate an anti-tumour response, as potentially does neutrophil directed therapy as described above.
There are examples where TAMs have been seen to elicit cancer destructive properties. Hagemann and colleagues have suggested that macrophages may be re-educated towards an anti-tumour function through control of NF-κB activity (Hagemann et al. 2008). Trabectedin is a recently approved chemotherapeutic that binds DNA and in so doing blocks the cell cycle affecting gene transcription and DNA repair. Importantly selected monocyte toxicity and inhibition of both CCL2 and IL-6 in vitro suggest a potential role in targeting macrophages in tumours (Germano et al. 2013). Three models of transplantable tumours, fibrosarcoma, lewis lung cancer, and ovarian carcinoma were assessed in vivo. There was a significant inhibition in each of these models of blood and splenic monocytes following trabectedin treatment. Furthermore, trabectedin treatment decreased tumour-associated macrophage numbers, a process dependent on caspase 8 activation. These findings highlight the potential for ablation of certain immunosuppressive cell types and the potential advantage of combinatorial approaches, for example anti-VEGF inhibitors whose efficacy is negated by TAMs. Measures to harness the immune system to eradicate tumour cells are being considered. An excellent example was recently provided in a cohort of metastatic PDAC patients who received CD40 targeting monoclonal antibodies in addition to gemcitabine (Beatty et al. 2011). CD40 activation, somewhat unexpectedly led to macrophage-dependent tumour regression. Promotion of properties of the adaptive immune system that protect us from malignancy in health holds promise for future trials.
Given recent evidence that immature myeloid cells referred to as BMDCs, classified as Gr1+, Cd11b+, Ly6C/Ly6G+ cells, have a profound effect on tumour immunosurveillance, and appear largely responsible for differentiation into both neutrophils (Ly6G+) and macrophages (Ly6C+), perhaps myeloid precursors themselves are the ideal targets for therapy in cancer. The progressive accumulation of BMDCs in PDAC is widely recognized, while their immunosuppressive effect is clear. They are known to have the capacity to differentiate into both neutrophils and macrophages (Kusmartsev et al. 2005), and it has been suggested that pro-tumourigenic N2 neutrophils (TANs) may differentiate from BMDCs of splenic origin. Indeed two groups have demonstrated that in response to oncogenic Kras dependent granulocyte, macrophage colony stimulating factor (GM-CSF) secretion, BMDCs are recruited to the tumour microenvironment where they play a role in suppressing CTL cell action and thus permitting tumourigenesis (Pylayeva-Gupta et al. 2012; Bayne et al. 2012). When GM-CSF was suppressed, BMDCs failed to infiltrate the tumour microenvironment and tumour growth was limited by infiltrating CTL cells. Furthermore, this effect was fully rescued by CTL depletion. Activated neutrophils also have the capacity to cause tissue damage via release of MMPs, while production of reactive oxygen species may promote mutagenesis. Reactive oxygen species have been implicated in the activation of NF-κB, which has been intimately associated with the inflammatory response and PDAC progression (DeNicola et al. 2011). Indeed the apparent importance of BMDCs in the mediation of immunosuppression and avoidance of surveillance raises their potential manipulation in concert with drugs that enhance T cell effectiveness permitting a CTL response. Regulation of T cell responses is mediated by both Cytotoxic T lymphocyte-associated antigen 4 (CTLA4) and PD1 checkpoints. Expression of ligands to these receptors on the surface of both myeloid cells and tumour cells can provide an off signal to the receptors and therefore failure to mount T cell response to cancer. Thus therapies that inhibit myeloid cell differentiation and chemotaxis to the tumour microenvironment are being combined with T cell activation strategies to attempt to overcome these complexities of the tumour microenvironment.
17.3.3.2 Stromal Signalling Pathways
In addition to immune cells other stromal elements play key roles in PDAC pathogenesis. Paracrine signalling through molecules including TGF-β, VEGF, hepatocyte growth factor, sonic hedgehog (Shh), epidermal growth factor, fibroblast growth factors and insulin-like growth factors all signal to the adjacent microenvironment, and in particular PSCs. This interaction activates and, is primarily responsible for fibrosis in PDAC (Neesse et al. 2011). They also implicated in local tumour growth in addition to their ability to travel to distant sites and support metastatic formation (Neesse et al. 2011). Elevated levels of factors secreted into the microenvironment including SPARC, a protein associated with cell migration and wound healing, have been associated with poor outcome in PDAC (Infante et al. 2007). Interestingly albumin-bound Paclitaxel (nab-paclitaxel) has been observed to bind SPARC-expressing fibroblasts, possibly providing a mechanism for targeting this specific cell type. Clinical trial of nab-paclitaxel in PDAC has shown some promise in combination with gemcitabine (Von Hoff et al. 2011), however the full role of SPARC in modulating the tumour microenvironment is unclear, with few changes seen in tumour stroma when preclinical PDAC models were treated (Frese et al. 2012).
The dense, avascular, collagenous extracellular matrix that constitutes the majority of the tumour bulk in human PDAC has been shown to mechanically block the vascular delivery of chemotherapeutic agents to tumours in preclinical models (Neesse et al. 2011). It is anticipated that drugs that attack stromal elements responsible for tumour maintenance will be of clinical benefit. Thus far, clinical trials assessing anti-stromal therapies including anti-MMP and VEGF inhibitors such as Bevacizumab have proven disappointing (Neesse et al. 2011). Among others, Shh signalling through Smoothened (Smo) has been implicated in the coordination of tumour-stromal crosstalk in PDAC. The clinical trial based on the work of Olive and colleagues (2009), who showed modest prolongation of survival of mice with PDAC following Smo inhibition in combination with gemcitabine, has been stopped due to better survival in the control arm (clinicaltrials.gov). However, there remains a belief that combating collagenous elements of PDAC stroma and promoting intra-tumoural vascularity will help delivery of standard chemotherapeutics to tumour cells. Tumours are dependent on both the protective effect of the stroma and interactions with stromal cells for progression. Hyaluronan has been identified as a key determinant of the stromal matrix with hyaluronidase treatment causing profound effects on tumour stroma in murine models of PDAC (Jacobetz et al. 2013; Provenzano et al. 2012). These drugs have also been shown to expand tumour vasculature and permit access of chemotherapeutics to the pancreas providing promise for ongoing clinical trials.
17.3.4 Emerging Importance of Inflammation in Metastasis
Metastasis, a hallmark of cancer, can be conceptualised as a multi-stage process where neoplastic cells spread from the tumour of origin and colonise distant sites. Local invasion, intravasation, survival in the circulation, extravasation and colonisation of the metastatic site are all required for development of metastases (Hanahan and Weinberg 2000). Therefore, the process of metastasis requires the coordination of a number of different biological processes. Pancreatic cancer sufferers die primarily as a result of tumour burden and commonly with metastatic disease despite surgical resection and adjuvant therapy. Indeed metastasis is the source of approximately 90 % of cancer deaths (Hanahan and Weinberg 2000) and is the major source of death in PDAC (seercancergov/statfacts/html/pancreas). In these patients, tumour cells either disseminate early in the course of PDAC, and colonisation takes longer to establish than primary tumours meaning these lesions are undetectable by pre-operative imaging, or, tumours disseminate late near to the point at which diagnosis is made and therefore colonisation goes undetected with current imaging modalities. There may be a period of colonization therefore, that allows a window of therapeutic opportunity following early diagnosis of the primary tumour.
The process of metastasis can be considered in the context of the ‘seed and soil’ hypothesis first proposed by Paget in 1889. This hypothesis suggests that microenvironmental elements are required by tumour cells (the seed) at distant sites (the soil) to permit establishment of metastatic disease.
Recent studies have shown metastases may occur in PDAC even before a primary tumour has formed confirming early dissemination, a behaviour associated with early epidermal mesenchymal transformation (Rhim et al. 2012). This process was accelerated in the presence of pancreatic inflammation, while the most invasive areas of tumour were seen at foci of inflammation. This phenotype was suppressed by dexamethasone, highlighting the integral role played by tumoural inflammation. NF-κB has been implicated in the process linking inflammation and cancer progression (Ling et al. 2012), however, it is likely that multiple different inflammatory pathways are implicated in this process including STAT-3 (Lesina et al. 2011), TLR4 signalling (Ochi et al. 2012b) and CXCR2 signalling (Ijichi et al. 2011).
‘Preconditioning’ of metastatic sites by non-cancerous immune cells including macrophages and BMDCs has been described (Barcellos-Hoff et al. 2013) in order to create a receptive soil for metastasis. It is thought that this pre-metastatic niche is established by such primary tumour components even prior to the presence of tumour cells at metastatic sites (Wels et al. 2008). BMDCs may cooperate with other molecules such as fibronectin, tenascin and MMP9 to develop a receptive environment for tumour establishment. It is believed that the production of chemokines and growth factors by primary tumour cells stimulate production of this environment. It has been suggested that CXCL1 production by breast cancer cells is important in metastasis formation in experimental models, implicating CXCR2 signalling in the establishment of the metastatic niche. However, full explanation of how CXCR2 signalling is implicated in the metastatic process in other cancers remains in its infancy. It is appreciated that primary lung cancer cells may produce VEGF-A, TGF-β, and TNFα resulting in upregulation of S100A8/9 in the premetastatic lung (Acharyya et al. 2012). S100A8/9 can be expressed by both neutrophils and macrophages (Passey et al. 1999). Furthermore it has recently been shown that progenitor BMDCs, particularly of the myeloid lineage, are responsible for upregulation of these molecules (Sade-Feldman et al. 2013). TLR4 signalling may also be important in the crosstalk between BMDCs and tumour cells during the process of metastatic niche formation (Barcellos-Hoff et al. 2013).
Macrophages have been described as ‘obligate partners for metastasis’ (Condeelis and Pollard 2006). Indeed genetic knockout of CSF1 in a mouse model of breast cancer almost completely abrogated metastases by interfering with macrophage homing (Lin et al. 2001). It has been shown that histidine-rich glycoprotein is important in suppressing the M2 protumourigenic phenotype of macrophages to a tumour inhibiting M1 phenotype. Blockade of placental growth factor was important for this phenotype. These effects suppress pro-angiogenic effects of macrophages and result in tumour vessel normalisation (Rolny et al. 2011). In addition vascular cell adhesion molecule (VCAM)-1 can tether breast cancer cells to tumour-associated macrophages during the development of lung metastases protecting the cells from immune cell destruction (Chen et al. 2011). These data provide an insight into the potential of tumour associated immune cells to influence cancer cell chemotaxis, invasion, and protection from immune destruction however, substantial investigation is required to clarify the underlying molecular mechanisms of these processes.
Extracellular components have been implicated in the process of metastasis. Lysyl oxidase (LOX) is a copper-dependent amine oxidase required for the covalent crosslinking of collagen. Significant associations between LOX and tumour progression have been observed, with high expression in breast, colorectal and head and neck cancers among others. In particular, LOX expression has been associated with hypoxia in tumors. Indeed in areas of hypoxia induction of LOX by hypoxia inducible factor has been observed to mediate metastases in vivo (Erler et al. 2006). Importantly LOX has been seen to play a critical role in pre-metastatic niche formation. LOX secretion by metastatic breast cancer cells cross-linked collagen IV to which BMDCs adhered releasing MMP2 to digest the matrix and allow influx of metastasising tumour cells (Erler et al. 2009).
PSCs are normally resident in the pancreas, and are responsible for the stromal reaction of PDAC. When quiescent they are characterised by vitamin A containing lipid droplets in their cytoplasm. These cells when activated lose this characteristic and become myofibroblast-like in morphology and function. They secrete an extracellular matrix component that forms fibrous tissue (Apte et al. 2004). There is crosstalk between PDAC cells and PSCs as has been demonstrated by the potential of PDAC cells to stimulate proliferation, migration and activity of PSCs in vitro (Bachem et al. 2005). Meanwhile PSCs are able to promote the survival of cancer cells through the inhibition of apoptosis (Vonlaufen et al. 2008). This highlights another way in which PDAC harnesses stromal cells to facilitate progression. PSCs are important in the development of the metastatic niche in PDAC. Using an orthotopic model of PDAC using human cancer cells mixed with human PSCs Xu et al. demonstrated that primary tumours were significantly larger and more stromal dense in the presence of PSCs (Xu et al. 2010). The metastatic rate was significantly higher in mice where PSCs were present within the orthotopic pancreatic injection group, and as PSCs were cultured from male patients and injected into female mice, fluorescence in situ hybridisation for the Y chromosome was able to demonstrate that PSCs from the primary site had indeed infiltrated secondary sites with the tumour cells. As yet few therapies have sought to target the signalling axis between PSCs and tumour cells.
17.4 Conclusions
In conclusion the myriad potentially activated inflammatory pathways across different cancers suggest that targeting of one pathway will seldom be sufficient to improve outcome, particularly in cancers such as PDAC. However, combination of a number of strategies, with chemotherapy may both improve chemotherapy efficacy and interfere with tumour-stromal crosstalk that mediates disease progression (Fig. 17.2). There are a number of well-characterised pathways through which the majority of tumoural inflammation is mediated. Many of these pathways have been shown to be important in tumour progression. Attractive targets include: general anti-inflammatories which target COX and other important inflammatory mediators; enhancement of cell mediated anti-tumoural immune response via reprogramming antigen presenting cells such as dendritic cells; activation of T cell mediated immune responses; removal of immunosuppressive BMDCs and macrophages from the tumour microenvironment; and interfering with commonly activated chemokines and transcription factors in pro-tumoural inflammation such as IL-6, TNFα, STAT3, and NF-κB. There is evidence from preclinical work that in combination with conventional chemotherapies these anti-inflammatory therapeutic approaches may activate scavenging effector systems as are seen in remodelling of wounds during healing (Coussens et al. 2013). At present there exists promising preclinical data regarding use of anti-inflammatory therapies in cancer. There are a few clear success stories such as the use of T cell checkpoint inhibitors in metastatic melanoma that provide hope for a large number of cancer patients in the future. However, only in the coming decade will we be able to judge the success of translation of these therapies from bench to bedside across different cancers. With such promising pre-clinical data, the time has now come for the translation to patient therapy with the aim of revolutionising cancer care and survival.
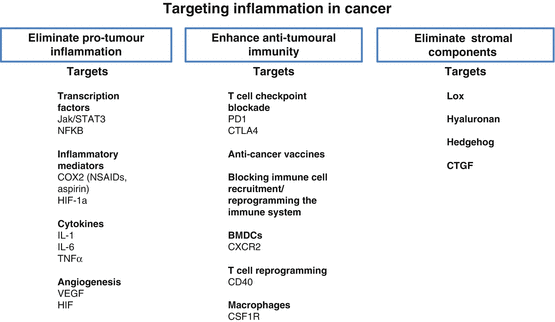
Fig. 17.2
 Introduction to Cell Stress Responses in Cancer: The Big Picture
Introduction to Cell Stress Responses in Cancer: The Big Picture
 Pro-senescence Therapy for Cancer: Time for the Clinic
Pro-senescence Therapy for Cancer: Time for the Clinic
 Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
 Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
 The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
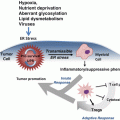 Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells
Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells




Targeting inflammation in cancer. Strategies to eliminate cancer via inflammatory modulation: Eliminate pro-tumoural inflammation; enhance anti-tumoural immunity; eliminate stromal components
Related posts:
Introduction to Cell Stress Responses in Cancer: The Big Picture
Pro-senescence Therapy for Cancer: Time for the Clinic
Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
