Acknowledgment
This work was supported, in part, by grants from National Institutes of Health (NIH), United States (R01 AI108907, R42AG054322, U54 AG062333, and R21 AG059742), and funding from Irma and Paul Milstein Program for Senior Health, Milstein Medical Asian American Partnership (MMAAP) Foundation, United States ( www.mmaapf.org ) to SXL.
Introduction
Osteoporosis and sarcopenia are two common age-related conditions well known to clinicians and researchers in geriatrics and gerontology. These two conditions often co-exist in an older and frail subset of the elderly population, giving rise to a new geriatric syndrome termed osteosarcopenia . While osteoporosis and sarcopenia are common and important clinical syndromes individually, osteosarcopenia has been increasingly recognized for its prevalence and significance as frail individuals are the fastest-growing subset of the older adult population. Osteosarcopenia entails significantly higher risks of physical functional impairment as measured by reduced grip strength and sit-to-stand power as well as chair rising time , falls, fractures, institutionalization , and mortality than osteoporosis or sarcopenia alone. These observations highlight the importance of advancing our understanding of the pathogenesis of this new geriatric syndrome and developing effective preventative and therapeutic interventions for it.
Muscle and bone are neighbors with close ties and crosstalk. They interact not only mechanically, but also through biological mediators and processes . Muscle and bone as an integrated unit represent a large portion of body mass and play a critical role in mobility and metabolic health in humans. At the same time, systemic biological processes, including metabolic disturbances, hormonal dysregulations, chronic inflammation, and aging, have a major impact on muscle and bone homeostasis. When such homeostasis is perturbed over time, it leads to the development of osteopenia/osteoporosis, sarcopenia, or both, i.e., osteosarcopenia. The shared or overlapping pathophysiology between osteoporosis and sarcopenia may serve as the pathophysiological basis for osteosarcopenia and point to exciting possibilities for potential interventions. As mechanobiology and a number of systemic biological processes are discussed elsewhere in the book, this chapter focuses on chronic inflammation. It will first provide an overview of age-related chronic low-grade inflammatory phenotype (CLIP) or inflammaging, termed CLIP-inflammaging. It will then present current evidence supporting the role of CLIP-inflammaging in contributing to osteoporosis, sarcopenia, and osteosarcopenia in older adults and discuss potential interventional strategies.
Age-related CLIP-inflammaging: Conceptual evolution and etiologic factors
Traditionally, inflammation, either acute or chronic, is regarded as a protective response that kills, inactivates, or walls off the insulting agent, such as an infectious agent or traumatic injury. Like many other immune reactions, this physiological response is highly regulated with prompt initiation and timely resolution . While too little or too late of this response would not provide effective protection, an uncontrolled or unresolved one would potentially lead to harm and pathologies.
CLIP, initially coined by Krabbe and colleagues , defines a low-grade, systemic, unresolved, and smoldering chronic inflammation clearly demonstrated by a two- to four-fold increase in circulating levels of inflammatory mediators such as interleukin-6 (IL-6) and C-reactive protein (CRP) in the absence of evident immediate triggers . “Inflammaging,” a term originally proposed by Claudio Franceschi , describes a chronic, systemic, low-grade inflammatory status that occurs during aging. Therefore, CLIP and inflammaging can be used interchangeably in the context of aging. As such, the two are combined that gives rise to the term CLIP-inflammaging. In addition to IL-6 and CRP, a large number of cellular (i.e., leukocytes and subpopulations, e.g., granulocytes and monocytes) and molecular inflammatory components are involved in CLIP-inflammaging or are produced as a result of inflammatory processes that lead to CLIP-inflammaging . For example, a plethora of pro- and antiinflammatory mediators have been described in the literature, including: (1) cytokines and their soluble receptors or associated molecules, such as IL-1, IL-1 receptor antagonist (IL-1ra), IL-6, IL-13, IL-18, interferon (IFN)-α and IFN-γ, tumor necrosis factor (TNF)-α and its soluble receptors 1 (sTNFR-1) and 2 (sTNFR-2); (2) chemokines including CCL2, CCL3, CCL5, IL-8, and CXCL10; (3) adhesion molecules, such as vascular cell adhesion molecule (VCAM)-1, intercellular adhesion molecule (ICAM)-1, and E-selectin; and (4) acute-phase reactants, such as CRP, serum amyloid A, and fibrinogen. This is not an exhaustive list and it continues to grow rapidly.
Etiologic factors and mechanisms of CLIP-inflammaging are likely multifaceted and remain to be fully elucidated. As a result, the concept itself continues to evolve. For example, “garb-aging” is a term in which endogenous, inappropriately exposed and misplaced, or misfolded or otherwise altered molecules or substances (collectively labeled as waste materials or “garbage”) were considered to be main sources or triggers of CLIP-inflammaging . These materials originate from damaged and/or dead cells accumulated during aging and are recognized by receptors of the innate immune system, suggesting an auto-inflammatory process . A related concept is “senescence-associated secretary phenotype” (SASP), suggesting that senescent cells can survive and accumulate in the circulation and tissues throughout the body during aging. While they no longer proliferate or perform much of their designated physiologic functions, they secret a wide range of pro-inflammatory mediators, thus contributing to CLIP-inflammaging . Another concept is “metainflammation,” which is literally translated as inflammation accompanying metabolic dysregulation, such as one associated with obesity, and suggests fat tissue as a major source of CLIP-inflammaging . Despite differences in terminology, these concepts emphasize multiple sources known to contribute to elevated levels of inflammatory biomediators in CLIP-inflammaging. Other potentially important etiologic factors that have been proposed include persistent viral infections, such as chronic cytomegalovirus (CMV) and human immune deficiency virus (HIV) infections, and dysregulated microbiota and gut permeability or gut dysbiosis . Finally, emerging evidence suggests defects in inflammation resolution, the final phase of any acute inflammatory response, as a potentially important mechanism for unresolved, persistent inflammation, leading to age-related development of CLIP-inflammaging . Understanding the key players in this active and dynamically regulated process will provide a basis for the potential development of effective interventional strategies for the prevention and treatment of CLIP-inflammaging, which will be further discussed later in this chapter.
CLIP-inflammaging: Clinical significance and potential underlying mechanisms
A large body of literature supports the notion that CLIP-inflammaging is a major risk factor for almost all age-related pathophysiological processes and chronic diseases, such as atherosclerosis and cardiovascular diseases, neurodegeneration and Alzheimer’s disease, insulin resistance and type 2 diabetes mellitus, tumorigenesis and cancer, as well as osteoporosis, sarcopenia, and frailty . The unique features of the clinical implication include (1) these chronic conditions are almost all age-related; and (2) they are otherwise seemingly unrelated pathophysiological processes and chronic conditions. The clinical significance is immediately evident because these chronic diseases are highly prevalent, often coexisting, and frequently lead to disability and early mortality in older adults.
Despite CLIP-inflammaging being recognized for the contribution to the aforementioned age-related pathophysiological processes and chronic diseases, the underlying mechanisms are far from being elucidated and likely context-dependent. One example is frailty, a common and important geriatric syndrome characterized by depletion of physiologic reserve and multiorgan system dysregulation. Substantial evidence suggests that CLIP-inflammaging contributes to the development of frailty in older adults directly and/or indirectly through other intermediate pathophysiologic processes . This is supported by a large number of studies documenting direct associations between frailty and CLIP-inflammaging as marked by increased counts of circulating white blood cells (WBC) and specific subpopulations (neutrophils, monocytes, CD8 + CD28 − and CCR5 + T lymphocytes) and elevated levels of molecular mediators of inflammation and immune activation (CRP, IL-6, TNF-α, neopterin, chemokine CXCL-10, etc.) . Moreover, studies have shown that circulating IL-6 levels have inverse associations with hemoglobin concentration and serum insulin-like growth factor-1 (IGF-1) levels only in frail older adults rather than in nonfrail controls, and WBC counts were inversely associated with IGF-1 levels; low hemoglobin and IGF-1 levels were each independently associated with frailty, as well. These and other studies suggest that CLIP-inflammaging may impact frailty through other intermediate pathophysiologic processes . In the context of cardiovascular diseases, atherosclerosis is regarded as a condition or pathophysiological process primarily caused by the accumulation of cholesterol-containing, oxidized LDL particles, seemingly unrelated to CLIP-inflammaging. However, oxidized LDL particles trigger an inflammatory response that can persist without timely resolution . Immune activation and subsequent CLIP-inflammaging can actively contribute to the initiation and progression of atherosclerosis, from early or further endothelial damage and dysfunction, acute thrombotic complications triggered by plaque rupture, to arterial wall fibrosis, calcification, stiffness, and stenosis, leading to the development of acute and chronic cardiovascular diseases. Therefore, CLIP-inflammaging contributes to the initiation and/or acceleration of this vicious cascade of events in the pathogenesis of atherosclerosis and cardiovascular diseases .
The following sections provide a brief summary of current evidence supportive of the role of CLIP-inflammaging in contributing to osteoporosis, sarcopenia, and osteosarcopenia.
Osteoimmunology and role of CLIP-inflammaging in contributing to osteoporosis
Osteoimmunology, initially coined by Aaron and Choi in the year 2000 , is an emerging interdisciplinary research field on the interactions between the bone and immune systems. These two seemingly unrelated systems not only have extensive reciprocal interactions, but also appear to be integrated with shared signaling pathways and regulatory mechanisms . A major focus of osteoimmunology is to investigate how the immune system impacts bone turnover in physiological and pathological conditions through the immunoskeletal interface . Understanding this interface will provide novel insights and help establish a framework for developing innovative preventative and therapeutic strategies for diseases related to both bone and immune systems.
Osteoporosis is a common geriatric syndrome characterized by loss of bone mass and bone mineral density (BMD) and disruption of bone microarchitecture to the extent that the skeleton becomes fragile, exposing the individual to increased risk of fractures . Osteoporosis is highly prevalent in postmenopausal women. As such, it was long considered simply as a result of postmenopausal estrogen decline. However, studies have shown that estrogen also has its receptor on immune cells, bone and bone marrow precursors, and that estrogen deficiency in postmenopausal women increases class II major histocompatibility complex human (MHCH) antigen expression on antigen-presenting cells (APCs) and inhibits T cell apoptosis, leading to T cell proliferation and activation. These effects result in the expansion of activated T cells, hyperproduction of pro-inflammatory cytokines, and chronic osteoclast stimulation, leading to bone loss and osteoporosis . Therefore, while postmenopausal estrogen decline is an important etiologic factor for osteoporosis, this estrogen deficiency may trigger or accelerate age-related CLIP-inflammaging, which, in turn, likely serves as the main underlying mechanism for bone tissue alteration and loss of BMD in postmenopausal women .
Substantial and growing evidence in the field of osteoimmunology indicates extensive sharing of signal transduction pathways and regulatory mechanisms between the bone and immune systems, which not only demonstrates resounding crosstalk and tight regulation between the two systems, but also firmly establishes the role of CLIP-inflammaging in the pathogenesis of osteoporosis with therapeutic implications . One key example is the receptor activator of nuclear factor-kB (RANK) and its ligand (RANKL). In the immune system, RANK/RANKL binding, through the adapter protein TNF-receptor-associated factor 6 (TRAF6), activates a number of key transcriptional factors, including nuclear factor 1 of activated T cells (NFATc1), activator proteins 1 (AP1), c-fos, and MAP kinases (MAPKs), the major hub of signaling pathways that govern immune activation and regulation . Importantly, RANK/RANKL also activates nuclear factor-κB (NF-κB) signaling, the canonical pathway for activation of the inflammatory cascade, leading to the production of a large number of pro- and antiinflammatory cytokines , many of which are main cytokine mediators involved in bone remodeling ( Table 1 ) . In the bone tissue, osteoblasts, osteocytes, and osteoclasts of the basic multicellular unit continuously communicate with each other through RANK/RANKL. For example, osteoblasts produce RANKL and macrophage colony-stimulating factor (M-CSF). The binding of RANKL to its receptor RANK on osteoclasts and their precursors is the main activation signal for bone resorption. This RANK/RANKL activation signal is further enabled by the binding of M-CSF and its receptor c-fms on these cells. Osteoprotegerin (OPG) inhibits osteoclastogenesis by acting as a decoy receptor of RANKL, thus preventing bone resorption . Therefore, RANK/RANKL is not only a crucial factor in immune regulation, but also the principle osteoclastogenic mediator . The clinical and translational implication of this mechanistic understanding is the development of an anti-RANKL monoclonal antibody, denosumab, as an effective therapy for osteoporosis with confirmed antiresorption activity of the bone .
| Cytokine | Proresorptive effects | Antiresorptive effects |
|---|---|---|
| IL-1 |
| |
| IL-4 |
|
|
| IL-6 |
|
|
| IL-7 |
|
|
| IL-8 |
|
|
| IL-10 |
|
|
| IL-11 |
|
|
| IL-12 |
|
|
| IL-13 |
|
|
| IL-15 |
| |
| IL-17 |
| |
| IL-18 |
| |
| IL-23 |
|
|
| IL-31 |
| |
| IL-32 |
|
|
| IL-33 |
|
|
| TNF-α |
| |
| TGF-β |
|
|
| INF-γ |
|
|
| SOFAT |
| |
| M-CSF |
| |
| G-CSF |
|
|
| HMGB1 |
|
The so-called secondary osteoporosis observed in patients with rheumatoid arthritis (RA) provides additional evidence supporting the role of inflammation in the pathogenesis of osteoporosis outside of the context of aging. In this autoimmune and inflammatory disease, increased bone resorption is a major pathophysiologic process leading to juxta-articular bone erosions, irreversible joint damage, and systemic osteoporosis, including decreased BMD in the spine and hip as well as a higher prevalence of fractures . Bisphosphonates, a class of potent inhibitors of osteoclast activity that are standard therapy for primary osteoporosis, are also effective for treating secondary osteoporosis in RA patients. Furthermore, monoclonal antibodies against various pro-inflammatory cytokines and their receptors, including those against TNF-α, are beneficial in preventing and reversing bone erosions and systemic osteoporosis in RA .
Of note, while most pro-inflammatory cytokines are osteoclastogenic mediators, there are cytokines that counteract bone resorption and exert osteoblastogenic properties, forming a complex bone remodeling cytokine network . Moreover, many cytokine mediators are pleiotropic and can exert different and, at times, contrasting effects, both pro-resorptive and antiresorptive effects, as listed in Table 1 . These pleiotropic effects largely depend on the specific context in which the cytokine mediators operate, the maturation stage of target cells, and the influence of other factors in the microenvironment, including cytokines. For example, in physiological bone remodeling, IFN-γ, a well-known pleiotropic cytokine, exerts antiosteoclastogenic effects by inducing TRAF6 proteasomal degradation with consequent inhibition of RANK/RNAKL signaling. However, in age-related, particularly postmenopausal osteoporosis, the overall effect of IFN-γ is skewed toward pro-osteoclastogenic/bone resorption through T-cell activation and RANKL expression. Studies have shown that IFN-γ is a powerful inducer of MHCH antigen expression on APCs, which, in turn, activates T cells along with specific antigen stimulation via T-cell receptors, inducing further immune activation and production of osteoclastogenic pro-inflammatory cytokines . These findings further suggest the level of complexity of this cytokine network as well as a heightened level of caution necessary in searching for therapeutic targets and designing interventional studies.
Role of CLIP-inflammaging in contributing to sarcopenia
Sarcopenia, defined by the loss of muscle mass and strength , is the key component or contributing factor of the syndrome of frailty , functional disability, and early mortality in older adults . An individual’s baseline muscle mass and function and their age-related rate of decline are influenced by one’s genetic background, i.e., the complex interplay of genetic and epigenetic factors . Major detrimental factors also include lack of exercise and physical inactivity, malnutrition, age-associated decline in systemic hormones (e.g., IGF-1, testosterone, and estrogen), alteration in muscle growth regulators (e.g., myostatin, etc.), and neuromuscular junction dysfunction .
Epidemiological studies have shown strong associations between CLIP-inflammaging and sarcopenia. For example, circulating levels of IL-6 and TNF-α are significantly elevated in older adults with sarcopenia . A large number of studies, including a meta-analysis, demonstrate a strong association of sarcopenia with increased circulating levels of various pro-inflammatory mediators, particularly CRP, IL-6, and soluble TNF-α receptors, compared to those in nonsarcopenic older individuals . In addition, systemic inflammation is considered as one of the primary mediators of skeletal muscle wasting in many chronic inflammatory conditions, such as RA, acquired immune deficiency syndrome (AIDS), chronic obstructive pulmonary disease (COPD), Crohn’s disease, and congestive heart failure . Moreover, data from longitudinal studies suggest that these associations may be causal. For instance, elevated serum IL-6 levels predict the development of sarcopenia, and high levels of IL6 and CRP at baseline are associated with a two- to -threefold greater risk of losing more than 40% of grip strength over 3 years . Furthermore, in a study that followed community-dwelling older women for 3.5 years, increased IL-6 levels were shown to predict a significant reduction of muscle strength and motor performance . Taken together, these and other longitudinal studies suggest that CLIP-inflammaging contributes, at least in part, to the onset and/or progression of age-related sarcopenia .
At the physiological level, CLIP-inflammaging impacts systemic hormones, such as IGF-1 and testosterone, major influencing factors of sarcopenia, similar to the associations between inflammatory mediators and IGF-1 levels in the syndrome of frailty cited above . At the molecular level, studies suggest that sarcopenia is associated with activation of catabolic pathways in the skeletal muscle governed mainly by three protein degradation systems, namely, the ubiquitin-proteasome system (UPS), the autophagy-lysosome system, and apoptosis. Activation of these protein degradation pathways leads to proteolysis, mitochondrial dysfunction, muscle fiber reorganization and denervation, and myofibril degeneration and myocyte death . On the anabolic side, mechanistic targeting of the rapamycin (mTOR) pathway is the key regulator of protein synthesis in skeletal muscle. Its activation increases protein translation, primarily myofibrillar proteins, and promotes transport of branched-chain amino acids (BCAAs) into the muscle cells whose adequate intracellular level is critical for continuous protein synthesis/recycling and expression of sirtuin 1 (SIRT1) and genes that are important for energy production. Taken together, activation of the mTOR pathway and its downstream molecules is important for stimulating cell growth and mitochondrial biogenesis as well as preventing “wear and tear” protein damage from accumulating, all of which facilitate muscle injury repair, prevent muscle atrophy, and maintain muscle quality and function . However, activation of the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome, typically triggered by cell debris and misfolded or damaged proteins, causes increased production of reactive oxygen species (ROS) and secretion of pro-inflammatory cytokines IL-1, IL18, and IL-33 , which in turn, can lead to not only activation of catabolic pathways, but also disturbed BCAA transport across cell membranes and impaired protein synthesis and mitochondrial biogenesis, resulting in muscle atrophy and dysfunction.
Perhaps the most direct evidence is from studies of animal models of acute inflammation. In rodents, inject of lipopolysaccharide (LPS), an endotoxin from Gram (−) bacteria, was shown to induce a systemic inflammatory response, increased expression of IL-6 and TNF-α mRNA in skeletal muscle, and activation of muscle catabolism through inflammatory signaling, resulting in loss of muscle mass and strength . Exogenous administration of TNF-α was shown to cause myofiber atrophy and muscle loss via several aforementioned mechanisms, such as activation of the canonical master regulator of inflammation pathway NF-κB, activation of UPS catabolic pathway, as well as a reduction in intracellular concentration of BCAAs and suppression of mTOR pathway of protein synthesis in the muscle . One interventional study in humans has demonstrated that blunting an acute infection-induced inflammation improved muscle performance in older adults . While limited translation potential of these findings from acute inflammation models to the setting of CLIP-inflammaging is recognized, it is conceivable that chronic elevation of these inflammatory mediators will likely adversely impact muscle mass and quality, leading to the development and/or progression of sarcopenia.
Role of CLIP-inflammaging in contributing to osteosarcopenia
As osteosarcopenia is a syndrome that encompasses both osteoporosis and sarcopenia, it is likely that CLIP-inflammaging contributes similarly to the underlying mechanisms, at least partially. Since osteosarcopenia is a recently defined syndrome, data from studies of osteosarcopenia as an integrated entity rather than its components are still scarce but rapidly growing. For example, the very first study of osteosarcopenia identified significantly higher circulating levels of uric acid, a well-established mediator of inflammation and endothelial dysfunction , in participants with osteosarcopenia compared to those with either of the two components or none .
Investigation into muscle, bone, and adipose tissue crosstalk, a recent line of research, may also provide novel insights into the role of CLIP-inflammaging in contributing to osteosarcopenia . Skeletal muscle, bone, and adipose tissue all produce cytokine mediators, namely, myokines, osteokines, and adipokines, respectively. These cytokine mediators exert autocrine/paracrine effects and establish a complex network that facilitates close interactions among the three seemingly unrelated organ/tissues. At the same time, many of these cytokine mediators have potent pro-inflammatory properties, exerting systemic effects as part of the CLIP-inflammaging. For instance, aging is associated with changes in body composition and increased adiposity. Adipocyte hypertrophy augments insulin resistance, perpetuating metabolic dysregulation. It recruits infiltrating immune cells, including macrophages that produce pro-inflammatory cytokines, further promoting CLIP-inflammaging . In addition, adipokines such as leptin upregulate IL-6 and TNF-α, and together downregulate adiponectin, the adipokine with antiinflammatory property. This metabolic and inflammatory vicious cycle of obesity not only promotes fatty infiltrates in the muscle tissue and bone marrow, but also feeds the catabolism and results in aberrant crosstalk of the bone, muscle, and fat tissue, initiating or exacerbating osteosarcopenia in older adults .
Tumor necrosis factor (TNF)-related weak inducer of apoptosis (TWEAK) is a pro-inflammatory and immune regulatory mediator that is increasingly recognized for its role in contributing to osteosarcopenia. TWEAK is a TNF superfamily member that is primarily produced by circulating immune cells, including resting and activated monocytes, dendritic cells (DCs), NK cells, as well as T and B lymphocytes . Its only known signaling receptor, fibroblast growth factor-inducible 14 (Fn14), is not expressed by these circulating immune cells, but mainly by epithelial cells, endothelial cells, and other cells in the periphery, including the skeletal muscle and bone. Fn14 is normally expressed at relatively low levels in healthy tissues, and TWEAK triggers its inducible expression. Therefore, the TWEAK/Fn14 pathway has been recognized as a critical “immune switch” that plays an important role in mediating the impact of inflammation and immune activation on the function and regulation of peripheral organ systems . For example, the TWEAK/Fn14 pathway may serve as an important underlying mechanism mediating the contributing role of CLIP-inflammaging to cardiovascular diseases and their severity. This is supported by the data from a clinical study of patients who were hospitalized for cardiovascular diseases in which TWEAK levels that were measured within 24 h after hospital admission were significantly higher in those with ST-elevation myocardial infarction (STEMI) than in healthy controls or patients with stable cardiovascular diseases . High TWEAK levels were also a predictor of adverse short-term outcome after STEMI . Several groups have also shown that TWEAK/Fn-14 pathway activation stimulates the production of pro-inflammatory cytokines and chemokines, such as IL-6, IL-17, matrix metalloproteinase (MMP)-3 and MMP-9, and R egulated upon A ctivation, N ormal T Cell E xpressed and presumably S ecreted (RANTES or CCL-5), triggering or perpetuating CLIP-inflammaging . In animal studies, genetic deletion of the gene encoding Fn14 was shown to be protective against myocardial hypertrophy, fibrosis, and heart failure; deletion of the gene encoding TWEAK or treatment with a TWEAK blocking antibody in atherosclerosis-prone mice resulted in the reduction of atherosclerosis burden and lesion size in the aorta with enhanced plaque stability , providing direct evidence supportive of the mediating role of the TWEAK/Fn14 pathway in contributing to cardiovascular diseases. Interestingly, recent data demonstrate that influenza immunization significantly reduced circulating TWEAK levels in older adults, particularly those who are frail, suggesting a potential underlying molecular mechanism for the observed cardiovascular protective effect of influenza immunization . While the TWEAK/Fn14 pathway may indirectly contribute to osteosarcopenia through the aforementioned cardiovascular pathologies with impaired blood supply, TWEAK has been shown to exert a direct detrimental impact on the skeletal muscle and bone. High TWEAK levels suppressed the self-renewal of progenitor cells and inhibited the differentiation of myoblasts. Upon injury and in disease conditions, TWEAK was observed to attenuate regeneration of damaged myofibers and promote fibrosis, leading to the impairment of skeletal muscle regeneration, differentiation, and repair, as well as its adequate structural and functional maintenance . The TWEAK/Fn14 pathway has also been recognized for its role in contributing to dysregulated bone remodeling. TWEAK/Fn14 activation was shown to inhibit osteoblast differentiation and promote its apoptosis . On the other hand, TWEAK/Fn14 was observed to induce RANKL expression, osteoclastogenesis, and poor bone mineralization, all of which lead to accelerated bone loss and destruction . Blocking TWEAK/Fn14 signaling either by monoclonal antibody against TWEAK or Fn14 or by Fn14-Fc suppressed expression of RANKL and IL-17 as well as osteoclast differentiation, providing further supportive evidence . Together, these studies suggest the important role of the TWEAK/Fn14 pathway in contributing to osteosarcopenia, both directly through its adverse impact on the skeletal muscle and bone and indirectly through its cardiovascular pathologies. The therapeutic implications are significant because not only biologicals specific to TWEAK and Fn14 for careful manipulation of the TWEAK/Fn14 signaling are available and appear to be safe in humans , but also TWEAK/Fn14 activities appear to be regulated by other immune interventions such as routine influenza vaccination . Vaccines are known to have nonspecific immunomodulatory effects .
CLIP-inflammaging in osteosarcopenia: Consideration of preventative and therapeutic interventions
Exercise and high levels of physical activities are believed to be the best nonpharmacological intervention with a wide range of health benefits for cardiovascular function and endurance, cognitive function, as well as metabolism and musculoskeletal system, and thus exercise is recommended for the prevention and treatment of almost all chronic conditions in older adults including osteosarcopenia . The importance of exercise and physical activity in maintaining musculoskeletal health and preventing osteosarcopenia in older adults is further emphasized by a 5-day bed rest study among well-characterized healthy young and old individuals, which demonstrates reduction of leg lean mass and strength along with increased markers of muscle proteolysis and osteoclastogenesis only in old participants .
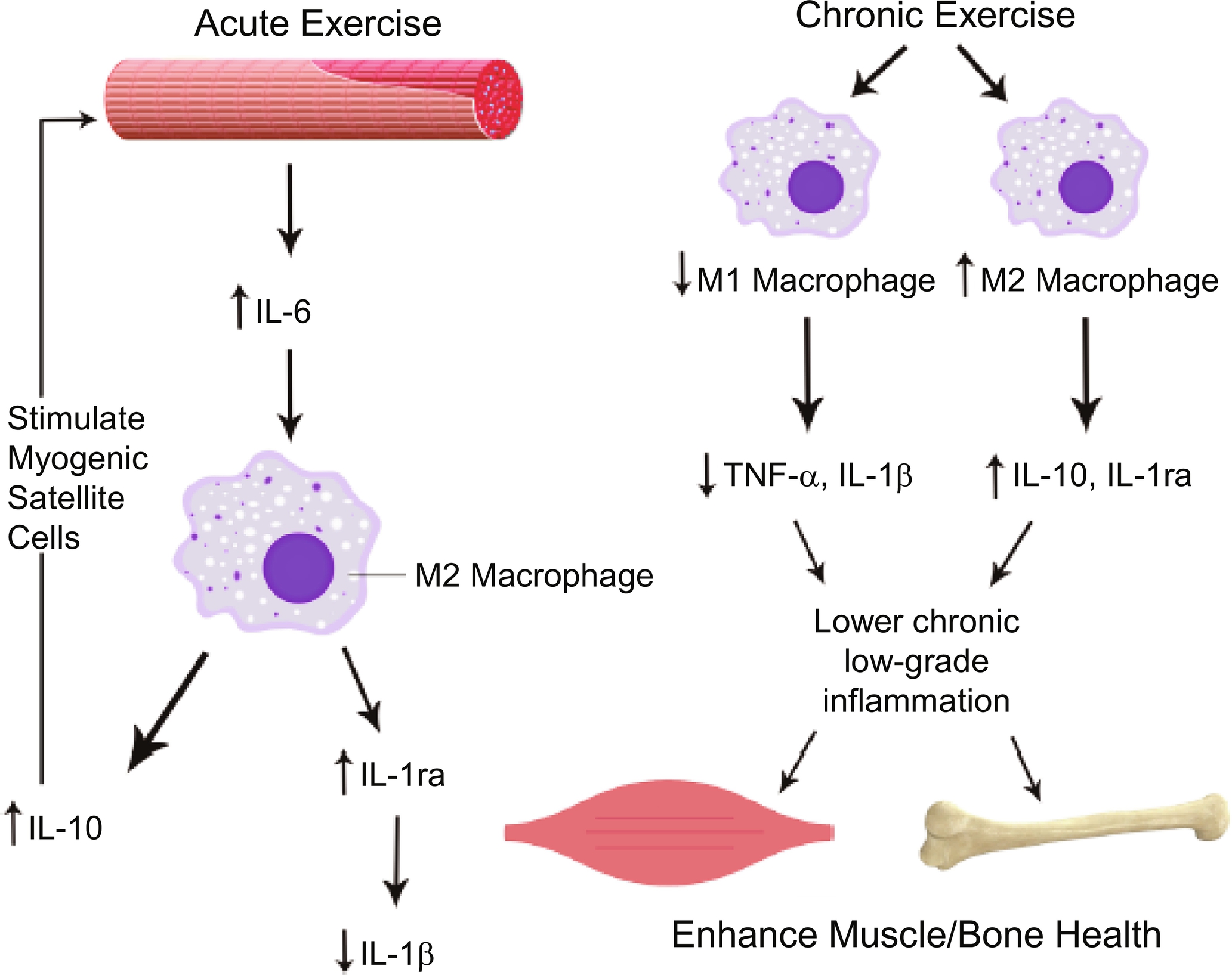
Exercise is also known to exert major positive impacts on the immune system . Substantial and growing evidence indicates that exercise imparts a potent antiinflammatory effect, which serves as an important mechanism underlying its preventative and therapeutic benefits for bone loss, sarcopenia, and osteosarcopenia . However, it is worth mentioning that an acute bout of exercise is known to induce a substantial increase in the production and circulating levels of IL-6, a seemingly paradoxical pro-inflammatory response . In this setting, IL-6, a pleiotropic cytokine mediator, is believed to reduce TNF-α production whose levels only increase locally within the skeletal muscle and remain undetectable or unchanged in the circulation . IL-6 also stimulates M2-type macrophages, the type of resident macrophages that are antiinflammatory in nature and involved in muscle regeneration, to produce antiinflammatory mediators, such as IL-10 and IL-1ra ( Fig. 1 ) . Regular (chronic) exercises, both endurance (aerobic) and resistance training, are known to suppress CLIP-inflammaging. While exercise reduces fat mass and pro-inflammatory adipokines, there is a significant phenotypic shift of resident macrophages in this setting, from the pro-inflammatory M1 type to the antiinflammatory M2 type, creating an antiinflammatory microenvironment within the skeletal muscle ( Fig. 1 ) . Consequently, exercise appears to exert its preventative and therapeutic effects on osteosarcopenia and other chronic geriatric conditions, at least in part, through its induced suppression of CLIP-inflammaging.
Related posts:
 Cellular senescence and other aging mechanisms in bone and muscle
Cellular senescence and other aging mechanisms in bone and muscle
 Biomechanics and mechanobiology of bone and muscle: Current and future musculoskeletal modeling research directions in osteosarcopenia
Biomechanics and mechanobiology of bone and muscle: Current and future musculoskeletal modeling research directions in osteosarcopenia
 Osteosarcopenic adiposity
Osteosarcopenic adiposity
 Sex steroids and gender differences in muscle, bone, and fat
Sex steroids and gender differences in muscle, bone, and fat
 Pharmacological management of osteosarcopenia
Pharmacological management of osteosarcopenia
 Frailty: The end of the osteosarcopenia continuum?
Frailty: The end of the osteosarcopenia continuum?
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


