and Steven D. Shapiro2
(1)
Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA, USA
(2)
Department of Medicine, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
Abstract
Numerous epidemiological studies have consistently linked the presence of chronic obstructive pulmonary disease (COPD) with increased lung cancer incidence even after correcting for cigarette smoke consumption. The mechanistic explanations for this epidemiologic link remain poorly understood as the two disease processes are seemingly polar opposites. However, there are key shared pathologic mechanisms that may link COPD and lung cancer, and inflammation is one of these factors, with the progression of both diseases substantially modified by the presence of chronic inflammatory cell infiltrates. Here, we review the most likely means by which cigarette smoke-induced inflammation would promote the development of lung cancer in COPD subjects.
Keywords
Lung carcinogenesisInflammationTumor microenvironment (TME)Epithelial–mesenchymal transition (EMT)Mesenchymal–epithelial transition (MET)Go versus grow hypothesisIntroduction
Despite recent research studies highlighting the ability of mutations in the epidermal growth factor receptor (EGFR) to instigate lung cancer in never smokers [1], smoking still remains the primary cause of approximately 90 % of all lung cancer cases [2]. The mortality statistics for lung cancer are staggering, accounting for nearly 160,000 deaths per year in the USA alone, and an estimated 1.3 million deaths each year worldwide [3]. Making matter worse, 5-year survival rates remain a dismal 15 %. Yet, the morbidity associated with lung cancer pales in comparison to that for chronic obstructive pulmonary disease (COPD) , which affects approximately 20 million people in the USA, where it has become the third leading cause of death annually [4]. As is the case with lung cancer, COPD is highly associated with cigarette smoking. The fact that both diseases share the same etiologic agent likely explains why it took decades for investigators to realize that these two diseases are actually linked. First reported in the mid-1980s, Skillrud [5] and Tockman [6] independently observed that the presence of COPD increased the incidence of lung cancer, even after controlling for cigarette smoke consumption. These findings have been reproduced in several independent investigations confirming that the presence of COPD increases the risk for lung cancer and lung cancer death [7].
The nature of the link between COPD and lung cancer remains obscure. Both of these diseases are mired by heterogeneous phenotypes and a substantial component of genetic susceptibility to disease. Lung cancer consists of multiple histologic subtypes, and COPD subjects display distinct clinical sub-phenotypes. At first glance, these diseases, in many ways, are polar opposites. Lung cancer is a disorder of uncontrolled cellular proliferation that displays enhanced angiogenesis . In contrast, COPD is a disease characterized by matrix destruction, cell death, and alveolar capillary dropout. A closer examination of the two disorders, however, will reveal key mechanistic links between COPD and lung cancer.
COPD is loosely defined as the presence of irreversible airflow obstruction (defined as a reduced ratio of forced expiratory volume in one second, FEV1.0, to total forced expiratory volume, or forced vital capacity, FVC) in the setting of chronic inhalation of particulate matter [8]. In the USA, this particulate matter is typically cigarette smoke, whereas it may be wood smoke or other biomass fuels in the developing world. COPD is typically subdivided into its two major components, the obstructive airways component, referred to as bronchitis, and the peripheral airspace disease, termed emphysema. It is important to note that both components almost always exist to some extent within a given subject. In contrast to the clinical definition of COPD , emphysema has a strict anatomic definition: the permanent enlargement of the peripheral airspaces of the lung distal to the terminal bronchioles [9]. There are two major theories with respect to the pathogenesis of emphysema. The proteinase–antiproteinase theory of emphysema pathogenesis still remains largely intact, now some 50 years after its inception [10]. It stems from the seminal observations of Laurell and Ericcson, who noted that three of the original five subjects with deficiency in alpha-one antitrypsin (A1AT), the physiological inhibitor of neutrophil elastase (NE), also had emphysema [11]. When instillation of the plant proteinase papain generated pathologic emphysema in the lungs of laboratory rodents, the concept was validated [12]. Although the exact type and source of the operative proteinase has changed over the years, the overall hypothesis remains more or less intact. Alternatively, more recent data support the notion that apoptosis of alveolar epithelial and endothelial cells drives the development of emphysema [13]. There is no question that programmed cell death is a central feature in cigarette smoke-induced emphysema, with the theory being supported by numerous studies. The major question that remains is whether cellular apoptosis is the primary event, or if it occurs downstream of inflammatory cell-mediated damage to the matrix scaffolding, which these cells need to survive.
Lung cancer arises from the accumulation of genetic mutations, likely a consequence of residing in an environment rich in carcinogens and oxidative stress [14]. Lung cancer is a heterogeneous disorder, with 20 % of the cases from neuroendocrine origin (small-cell lung cancer, SCLC), and the other 80 % being lumped together as non-small cell lung cancer (NSCLC) [15]. NSCLC comprises mostly adenocarcinoma (ADCA) and squamous cell carcinoma (SCCA), though there exist other less common subtypes, such as large cell carcinoma (LCC). Both lung ADCA and SCCA (as well as SCLC) are linked with COPD, though there are insufficient data with respect to the other less common subtypes.
The most glaring link between COPD/emphysema and lung cancer is inflammation . Essentially all habitual cigarette smokers will develop macrophage and neutrophil-rich pulmonary inflammation. Here, we will explore specific roles for inflammatory cells in COPD and lung cancer, in attempts to identify unique mechanistic links.
Radiographic Emphysema Confers Cancer Risk
Little progress was made during the two decades following Skillrud’s initial report that COPD and lung cancer were linked. Several other large series were able to reproduce the findings in independent cohorts, such that the epidemiological data supporting the link are very strong (Table 1.1) [16–19]. However, it was not until researchers began the process of sub-phenotyping COPD subjects that novel findings were presented. Using CT scans to measure the amount of radiographic emphysema, researchers began to characterize the COPD disease state as a combination of airspace disease (CT) and airflow obstruction (pulmonary function tests, PFTs). Using a cohort defined in this way, de Torres and colleagues queried whether the presence of airflow obstruction or radiographic emphysema conferred lung cancer risk [20]. Their results showed that the presence of radiographic emphysema, and not airflow obstruction per se, conferred cancer risk. These findings have been reproduced in a number of independent studies [21–23] with some caveats. First, all of the other studies have found that both radiographic emphysema and airflow obstruction confer cancer risk, such that both airspace and airways disease are involved. Second, the methods employed by researchers to measure radiographic emphysema considerably affect the results. Initially, investigators used a semi-quantitative assessment of emphysema that was dependent upon the investigator. Subsequently, computer-generated algorithms were created to quantitatively measure the presence of emphysema. Studies employing this methodology have failed to reproduce the original results [24], which may be related to the inability of the software to detect the presence of mild emphysema. Highlighting the importance of these methodological issues, two studies out of the University of Pittsburgh utilizing the exact same patients and scans obtained opposite results when comparing quantitative and semi-quantitative measures of radiographic emphysema [21, 25]. Further study will likely be required to determine the optimal means to measure radiographic emphysema in this setting.
Table 1.1
COPD is linked to lung cancer incidence
Study | N | Outcome | FEV1 (% predicted) | Emphysema |
|---|---|---|---|---|
Skillrud | 226 | Incidence | Cancers in 8.8 % of cases (FEV1 < 70 %) vs. 2.0 % of controls (FEV1 > 85 %), p = 0.024 | N/A |
Tockman | 4395 | Mortality | Cohort 1, RR 4.85 for FEV1 < 60 % vs. > 60 %, p = 0.002 Cohort 2, RR 2.72 for FEV1 60–85 % vs. > 85 %, p = 0.043 | N/A |
Speizer | 8427 | Mortality | Quartile based FEV1 analysis confers cancer risk (RR 2.0–8.27) | N/A |
Lange | 13946 | Mortality | RR 2.1 (95 % CI 1.3–3.4) for FEV1 40–79 % vs. > 80 %; RR 3.9 (95 % CI 2.2–7.2) for FEV1 < 40 % vs. > 80 % | N/A |
de Torres | 1166 | Incidence | RR 2.89 (95 % CI 1.14–7.27) for FEV1/FVC ratio < 70 % vs. > 70 % | Semi-quantitative radiographic emphysema, RR 3.13 (95 % CI 1.32–7.44) |
Wilson | 3638 | Incidence | OR 2.09 (95 % CI 1.33–3.27) for any GOLD stage (FEV1/FVC < 70 %) | Semi-quantitative radiographic emphysema, OR 3.56 (95 % CI 2.21–5.73). After controlling for airflow obstruction, OR 3.14 (95 % CI 1.91–5.15) for radiographic emphysema |
Li | 1015 | Incidence | N/A | Semi-quantitative radiographic emphysema, Any = OR 2.79 (95 % CI 2.05–3.81), > 5 % = 3.80 (95 % CI 2.78–5.19), > 10 % = OR 3.33 (95 % CI 2.30–4.82) |
Zulueta | Mortality | N/A | Semi-quantitative radiographic emphysema, HR 1.7 (95 % CI 1.1–2.5), p = 0.013 | |
Maldanado | Incidence | Cancer risk conferred by decreasing FEV1, OR 1.15 (95 % CI 1.00–1.32, p = 0.046); and FEV1/FVC < 70 %, OR 1.29 (95 % CI 1.02–1.62, p = 0.0310) | Automated volumetric determination of radiographic emphysema was not associated with lung cancer risk, OR 1.042 (95 % CI, 0.816–1.329, p = 0.743) |
Inflammation as the Link Between COPD and Lung Cancer
The association between inflammation and cancer is not new, dating to classic observations made by the anatomist Galen in the second century A.D., when he postulated the Greek version of the famous four symptoms of inflammation : rubor, calor, dolor, and tumor [26]. Even the more modern concept that tumors arise in areas of chronic inflammation is not as modern as one would think, having been discovered by Virchow in 1863 [27]. Although Virchow’s hypothesis remains intact, it is only recently that specific components of the immune system have been demonstrated to play vital roles in tumorigenesis beyond creating a chronic inflammatory milieu in which neoplastic transformation could occur.
There is no question that inflammation is common to both lung cancer and COPD/emphysema. Inflammatory cells are readily present in lung cancer histology sections of all subtypes [28]. Early studies employing bronchoalveolar lavage fluid (BALF) analysis to study the inflammatory cell content in smokers with and without COPD showed a uniform increase in macrophages and neutrophils in the lungs of smokers [29, 30]. Although always present, the severity of inflammation is greater in smokers with COPD than those without. Furthermore, inflammatory cell content tends to correlate with disease severity in COPD .
Questions arise when one considers the exact cellular types and subtypes present in COPD/emphysema and lung cancer subjects (Fig. 1.1). A landmark study by Hogg and colleagues demonstrated that the quantity of CD4 + and CD8 + T lymphocytes as well as B-lymphocytes within COPD airways correlated with disease severity [31]. Yet, the subtypes of CD4 + lymphocytes in COPD are still poorly understood. Since emphysema is a destructive disease, it is logical to assume that the immune cells present in COPD lungs would be cytotoxic in nature. Accordingly, work in humans and mice suggests that COPD/emphysema represents a Th1 disease state. The (interferon-γ) IFNγ elaborated from Th1 cells (CD45 + CD4 + IFNg +) is thought to elicit a response from the IFNγ-inducible genes, such as CXCL-9, CXCL-10, and CXCL-11 [32]. CXCL-10 (also known as IFNγ-inducible protein-10, IP-10) elaborated from CD8 + lymphocytes and epithelial cells has been shown to stimulate the release of matrix metalloproteinase-12 (MMP-12) from macrophages [33], which is well known to cleave elastic fibers [34], a hallmark of emphysema pathogenesis. The recruitment of neutrophils in this setting is likely due to the release of IL-8 from alveolar macrophages and neighboring epithelial cells [35]. There are some gaps in this schema, however. A predominance of Th1 cells in COPD subjects has never been explicitly documented. The data most supportive of Th1 infiltration used individual STAT4 and IFNγ stains on airways [36]. Without the use of multiple markers, it is possible that other immune cell types (macrophages and CD8 + lymphocytes, for example) have been mistaken for Th1 lymphocytes. Additionally, the macrophages in COPD are typically of a mixed M1/M2 phenotype [37, 38], though macrophages release matrix-degrading proteinases rather uniformly, independent of their cellular phenotype. Nonetheless, the overarching theme is sound, that the cells present within emphysematous lungs are cytotoxic and destroy extracellular matrix.
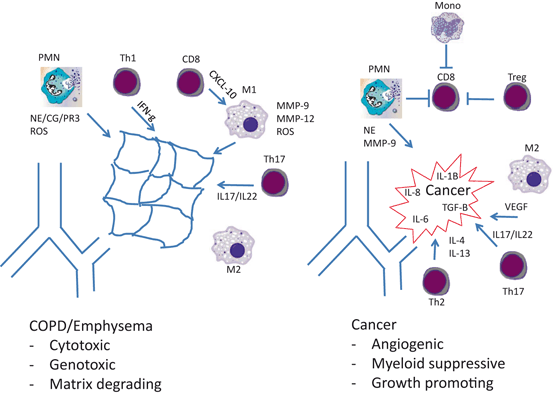
Fig. 1.1
Immune cell content in COPD and lung cancer. COPD chronic obstructive pulmonary disease, PMN polymorphonuclear leukocytes, MMP matrix metalloproteinase, NE neutrophil elastase, CG cathepsin G, PR3 proteinase-3, ROS reactive oxygen species, IFN interferon, VEGF vascular endothelial growth factor, TGF-B transforming growth factor beta
Unfortunately, it does not appear that tumor-associated inflammation is cytotoxic. Specifically, the CD8 + cells present within the tumor microenvironment (TME) are notoriously dysfunctional, likely the result of hyperactive checkpoint regulation, designed to prevent autoimmune disease [39, 40]. Recent clinical trials in NSCLC subjects have highlighted the potential benefit of having functional tumor infiltrating cytotoxic T lymphocytes (CTL). The administration of antibodies against key checkpoint regulation targets (e.g., PDL1) was able to restore CD8 + function, which resulted in sustained tumor regression in approximately 20 % of these NSCLC subjects [41]. A better understanding of the mechanisms contributing to CTL dysfunction in cancer subjects should provide additional therapeutic targets to optimize CD8 + function within the TME.
Although Th1 lymphocytes are identifiable within tumors, the TME is best characterized as a Th2, or wound-healing environment [42]. Th2 lymphocytes, characterized by IL-4 and IL-13 expression, tend to drive alternatively activated macrophages , deemed M2. Indeed, tumor-associated macrophages encountered at sites of tumorigenesis are typically of this wound healing, or M2 variety. These cells are highly angiogenic and believed to greatly assist developing tumors in this regard. Additionally, macrophages release cytokines and chemokines to maintain tumor-promoting inflammatory cell infiltrates, as well as other key growth factors [43].
Neutrophils are an undoubtedly common link to both COPD/emphysema and lung cancer. As discussed above, essentially all habitual cigarette smokers develop some level of neutrophil accumulation within the lungs. More recent evidence has demonstrated that polymorphonuclear leukocytes (PMN) are present at sites of tumorigenesis as well [44]. In some cases, the existence of tumor-associated PMN appears to be the result of a tumor-driven process. For example, mutant K-ras directly induces the expression of IL-8 in an NFkB-dependent process [45]. Therefore, the presence of tumor-infiltrating neutrophils in Lox-Stop-Lox K-rasG12D (LSL-K-ras) mice is the result of tumor behavior, and likely does not represent a means of host defense [46]. Accordingly, recent studies in human NSCLC tissues have confirmed that neutrophil content in tumors inversely correlates with outcomes [47]. The exact mechanisms by which PMN are recruited to K-ras WT lung cancers remain a work in progress.
There is mounting evidence to support a role for the more recently described CD4 + lymphocyte subsets in COPD and lung cancer. Th17 lymphocytes, deemed as such because they produce the largely site-restricted cytokine IL-17, are pro-inflammatory cells known to propagate chronic inflammation [48]. Their role in chronic inflammatory disorders, such as rheumatoid arthritis, is well documented [49]. IL-17 serves to amplify the release of CC and CXC chemokines, which serve to recruit monocyte/macrophages and neutrophils, respectively [50]. Since COPD is a chronic inflammatory disease, and lung cancers have been likened to wounds that would not heal, investigators have begun to query whether Th17 lymphocytes are responsible for this chronic inflammation . The initial reports are supportive. Cigarette smoke exposure in and of itself increases the numbers of IL-17-producing cells in the lungs. This is a result of aryl hydrocarbon receptor (present on CD4 + cells) interaction with cigarette smoke constituents that directly results in IL-17 expression. Exposure of IL-17 receptor A-deficient mice (IL-17RA−/−) to cigarette smoke demonstrated an important role for IL-17, as these mice were protected from the development of emphysema and macrophage accumulation [51]. Not surprisingly, CC chemokine production was inhibited in this setting. An additional source of IL-17 production within the lungs is the γδ T cell, a unique cell population at the interface of innate and adaptive immunity. Cigarette smoke exposure experiments in mice have documented an instrumental role for this lymphocyte subset as well [52].
Initial reports regarding the role of IL-17 in tumor growth were contradictory [53, 54]. More recent reports support a pro-tumor role for these cells in solid tumor growth. Although the mechanisms have yet to be fully elucidated, there is evidence to support a role for Th17 cells in the propagation of myeloid cell infiltration and the ability of developing cancers to form an adequate vascular supply [55].
Oxidative and Toxic Stress is Common to COPD and Lung Cancer
The lungs of chronic cigarette smokers are bombarded with toxic and oxidative stressors. In this respect, the constituents of cigarette smoke, and enzymes housed within inflammatory cells, work in concert to provide this stress. Cigarette smoke provides substantial oxidative stress in and of itself, but only a fraction of the burden generated by the influx of macrophages and neutrophils, both of which are laden with reactive oxygen species (ROS)-producing machinery. Inflammatory cells are also capable of generating unique ROS types that would not be delivered directly by smoking cigarettes. Specifically, the neutrophil enzyme myeloperoxidase (MPO) is responsible for the generation of hypochlorous acid (HOCL–) [56]. Ultimately, such oxidative stress presents two fundamental problems for lung epithelial cells residing in such an environment: genotoxic and apoptotic stress. To combat oxidative stress, epithelial cells possess a set of genes, such as catalase, which contain an antioxidant response element (ARE), regulated by nuclear factor erythroid 2-related factor 2 (Nrf2) [57]. The initial reports with respect to Nrf2 function in emphysema were quite impressive. Nrf2−/− mice develop a greater amount of cigarette smoke-induced emphysema when compared to control mice [58]. Mechanistically, there was a greater amount of epithelial cell apoptosis in the absence of Nrf2, which resulted in enhanced airspace disease. Such findings led to the development of Nrf2 agonists [59, 60], with the assumption that reducing the oxidative stress in the lungs of cigarette smokers would be beneficial in both COPD and lung cancer. Unfortunately, studies examining the role of Nrf2 in cancer have provided conflicting results. The literature shows clear evidence of both pro-tumor and pro-host functions of Nrf2 in solid tumor models [61, 62]. Specifically concerning has been the revelation that Nrf2 activity can serve to metabolize chemotherapeutic agents, thereby reducing the effectiveness of anti-cancer therapies [63]. Although there may yet prove to be effective strategies involving Nrf2 agonists, such interventions would have to be very context specific.
There exist two additional aspects of cigarette smoke that present challenges to smokers in addition to oxidative stress. First are simply the addictive components of smoke, such as nicotine, that ensure chronic usage [64]. Second are the numerous toxic and carcinogenic agents found among the ~ 4000 distinct chemical species housed within each puff. Of numerous potential candidates, data are strongest to support pathogenic roles for carbon monoxide, N-nitroso derivatives, polycyclic aromatic hydrocarbons (PAHs), and 4-methylnitrosamino-1-[3-pyridyl]-1-butanone (NNK). The detoxification system handles such insults by using a two-step or two-phase system to combat such substances. In the first step, phase I enzymes such as heme oxygenase 1 (HO1), MPO, and members of the epoxide hydrolase (EPHX) and cytochrome P450 (CYP) families function to activate the substrate via oxidation, reduction, or hydrolysis [65]. The second step, performed by phase II enzymes (e.g., GSTs), is the conjugation of the agent, which allows for excretion. These systems are generally effective in detoxifying carcinogens found in cigarette smoke, with a few notable exceptions. One of the best-known cigarette smoke-derived carcinogens is the PAH benzo-a-pyrene (B(a)P), which in and of itself is not actually a carcinogen. B(a)P is only carcinogenic after it is converted to metabolites by EPHX1 [66]. The function of these enzymes is not trivial, as functional SNPs within the phase I system significantly alter cancer risk, most notably for MPO [67].
Proteinases Contribute to Lung Matrix Destruction and Tumor Growth
There is extensive literature documenting roles for extracellular matrix degrading proteinases in the pathogenesis of both lung cancer and emphysema (Table 1.2) [68]. Such roles for these enzymes were established much earlier in emphysema than in lung cancer, owing to the proteinase–antiproteinase hypothesis described above. Since that time, key roles for members of the cysteine (cathepsins), metallo- (matrix metalloproteinase, MMP), and a disintegrin and metalloproteinase domain (ADAM) and serine proteinase families have been documented in almost all aspects of biology, ranging from development to cancer [69]. With respect to emphysema, it is believed that the presence of elastolytic activity is prerequisite for a proteinase to contribute significantly to disease pathogenesis. However, there are exceptions, with MMP-1 being the most notable. For that reason, the emphasis here will be placed on elastolytic proteinases that also promote tumor growth. Most of these enzymes originate from macrophages , neutrophils, and inflammatory monocytes, although fibroblasts almost certainly contribute. Since macrophages and neutrophils are ubiquitously present in emphysema and lung cancer, the presence of many of these proteinases is a near certainty. Importantly, all of these enzymes are inhibitable, and therefore therapeutically relevant. Unfortunately, the initial attempts to target these enzymes in cancer were premature, and took place before detailed experimental data were available. Such studies, performed later, provided valuable insights with respect to the presence of both pro-host and pro-tumor roles for these enzymes in cancer.
Table 1.2
 Inflammation and Lung Cancer: Molecular Pathology
Inflammation and Lung Cancer: The Role of Epithelial–Mesenchymal Transition
Inflammation and Lung Cancer: Addressing Inflammation with Immunotherapy
Inflammation and Lung Cancer: Molecular Pathology
Inflammation and Lung Cancer: The Role of Epithelial–Mesenchymal Transition
Inflammation and Lung Cancer: Addressing Inflammation with Immunotherapy
 Inflammation and Lung Cancer: The Link to Angiogenesis
Inflammation and Lung Cancer: Prevention
Inflammation and Lung Cancer: The Link to Angiogenesis
Inflammation and Lung Cancer: Prevention
 Inflammation and Lung Cancer: Eicosanoids
Inflammation and Lung Cancer: Eicosanoids




Proteinases operative in COPD and lung cancer
Related posts:
Inflammation and Lung Cancer: Molecular Pathology
Inflammation and Lung Cancer: The Role of Epithelial–Mesenchymal Transition
Inflammation and Lung Cancer: Addressing Inflammation with Immunotherapy
Inflammation and Lung Cancer: The Link to Angiogenesis
Inflammation and Lung Cancer: Prevention
Inflammation and Lung Cancer: Eicosanoids
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
