Potential linkers
Source
Functions in inflammation
Functions in angiogenesis
References
TNF-α
Macrophages
Tumor cells
Augments E-selectin and VCAM-1 on ECs to promote leukocyte adhesion and inflammation
Induces COX-2
Induces IL-8
Induces expression of VEGF and HIF-1α in tumor cells
bFGF (FGF-2)
ECs
Fibroblasts
Tumor cells
Enhances leukocyte recruitment and EC adhesion molecule (CAM) expression during inflammation
EC proliferation
EC migration
Induces VEGF in ECs
Ang-1
Mesenchymal cells
Pericytes
Suppresses inflammatory gene expression
Stabilizes nascent vessels by activating Tie2 on endothelial cells
Ang-2
ECs
β2-integrin-dependent recruitment of myeloid cells
Ang-2 promotes vascular sprouting and destabilizes blood vessels by disrupting interactions between ECs and peri-ECs
HIF-1α
Hypoxic tumor cells
Macrophages and neutrophils
Regulates the metabolic switch to glycolysis in myeloid cells, allowing their infiltration into hypoxic inflammatory sites
Induces VEGF
COX-2
Macrophages
ECs
Tumor cells
PGE2 downstream of COX-2 increases local blood flow
Upregulates VEGF in tumor cells
Induces HIF-1α
IL-1β
Macrophages
Tumor cells
Induces COX-2.
Induces IL-8
Induces VEGF in aortic smooth muscle cells
Upregulates HIF-1α
IL-6
Macrophages
Tumor cells
Activates endothelial cells secretion of IL-8 and MCP-1, recruiting myeloid cells
B cell differentiation
T cell activation
Promotes expression of VEGF through JAK2/STAT3 signaling
IL-8 (CXCL8/CXCL2)
Neutrophils
Tumor cells
Macrophages
Recruits neutrophils
Induces neutrophil respiratory burst
Enhances EC survival, proliferation, and MMP-2 and MMP-9 expression
VEGF-A
EC
Macrophages
Amplifies inflammation, by acting as chemoattractant for myeloid cells.
Enhances TNFα-induced monocyte recruitment and P-selectin, E-selectin, and ICAM-1 expression
EC survival
EC proliferation
EC migration
Tube formation
CXCL12 (SDF-1)
Perivascular myofibroblasts
Attracts myeloid cells
Retains myeloid cells in close proximity to angiogenic vessels, promoting endothelial cell proliferation
[58]
MMPs
Neutrophils
Promote diapedesis by degrading ECM
Increase VEGF bioavailability by releasing it from ECM
Along with the VEGF–VEGFR system, the angiopoietin (Ang)–Tie ligand–receptor system plays an important role, as it not only regulates angiogenesis but also controls endothelial inflammation [44, 72]. Ang-1 is secreted by pericytes and activates the tyrosine kinase receptor, Tie2, expressed on ECs. This signal stabilizes nascent vessels, inhibits vascular leakage, and suppresses inflammatory and coagulation-related gene expression [15, 44]. As such, Ang-1 can be considered a paracrine switch that controls the transition from an inflammatory or angiogenic endothelium to a resting endothelium. Ang-2, on the other hand, is thought to be an antagonist of the Tie2 receptor and blocks the action of Ang-1. Notably, Ang-2-deficient mice displayed defects in the generation of rapid inflammatory responses, and the state of Ang-2 deficiency influenced leukocyte adhesion and transmigration [4, 72] (Fig. 5.1).
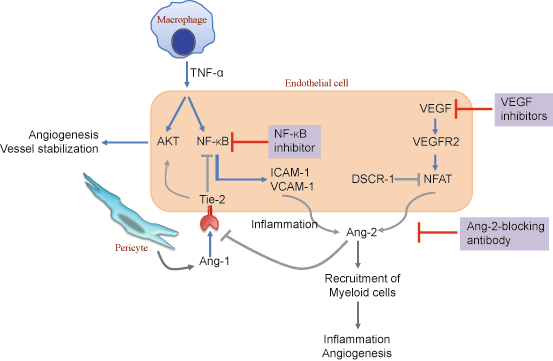
However, for a long time it had remained uncertain whether the angiogenic signals are direct regulators of the inflammatory process, or simple mediators that merely propagate and/or amplify other preceding proinflammatory stimuli. Notably, a recent study in mouse has demonstrated that Ang-2 expression in ECs is sufficient to recruit myeloid cells in a β2-integrin dependent manner to induce inflammation even in the absence of preceding proinflammatory stimuli [144]. In line with this observation, Ang-2-blocking antibody inhibited tumor growth and metastasis by impairing angiogenesis and disrupting the recruitment of pro-angiogenic myeloid cells [106].
The concept of Ang-2 as a proinflammatory agent has important clinical implications, considering the intimate relationship of inflammation with various diseases, including lung cancer and metastasis. In this regard, serum Ang-2 levels were found to be significantly higher in lung cancer patients, and positive correlations were observed between serum Ang-2 and tumor stage and metastasis [117, 127]. Notably, Ang-2-blocking antibody inhibited lung metastasis in part by enhancing the integrity of EC–EC junctions in mice [67]. In a recent study, Calcineurin-NFAT was observed to be activated in the lung endothelium of the pre-metastatic niche, which in turn upregulated Ang-2 to promote the angiogenic switch, resulting in increased lung metastases [114]. Together, these studies are likely to have implications towards the clinical application of Ang-2-modulating therapies.
Cellular and Molecular Regulators Linking Inflammation and Angiogenesis to Lung Cancer
The lung is a specialized organ of the respiratory system and is composed of extensive vascular beds. The stroma underlying the parenchyma of the lower airways is highly vascularized, and is generally subjected to higher oxygen tension [128]. Compared to other organs, neoangiogenesis is not apparent in the lungs since the vascular bed is extensive and fully developed [62]. However, as we will discuss later, the ECs play an active role in tissue remodeling in pathological conditions. Furthermore, the presence of an extensive endothelial surface makes the lungs prone to infiltration by circulating immune cells [9]. In some cases, persistent hyperactivation of inflammatory cells results in pathological manifestations such as emphysema, COPD, or interstitial lung disease (ILD) [91, 166].
The lung vasculature is mainly comprised of capillaries that line the alveolar walls, to facilitate gaseous exchange [150]. In addition to the capillaries, larger vessels such as the pulmonary artery and veins provide considerable endothelial heterogeneity. Studies in animal models have shown that the inhibition of angiogenesis in embryonic lung results in vascular defects associated with a significant reduction in the formation of air space and capillaries, which results in distended and underdeveloped alveoli [30]. The vascular bed in the adult lung is generally quiescent, and the equilibrium between quiescence and activation is exquisitely controlled by pro- and anti-angiogenic molecules [113]. While a balance of these factors maintains stability of the vasculature, overproduction of pro-angiogenic factors, or other imbalances in this equilibrium leads to aberrant blood vessel development.
Inflammatory tissues are often hypoxic, and hypoxia stimulates angiogenesis by virtue of hypoxia-inducible factor-1 (HIF-1)-mediated VEGF production [53, 74], which activates ECs [133]. It has also been shown that inflammatory cytokines, such as IL-1β, can upregulate HIF-1α through a classical inflammatory signaling pathway involving NF-κB and COX-2, culminating in upregulation of VEGF, a potent angiogenic factor required for tumor growth and metastasis [78]. Thus, HIF-1 has been implicated as a pivotal transcription factor linking the inflammatory and oncogenic pathways.
Among the inflammatory cells, myeloid cells that comprise a major component of the leukocyte pool are key regulators of angiogenesis. Particularly, neutrophils, monocytes, basophils, and mast cells secrete angiogenic factors that induce the angiogenic switch [26]. Neutrophils play an important role in the immune response to lung cancer as well as COPD. A recent study showed a higher degree of local and systemic neutrophilic inflammation in patients with lung cancer (with or without COPD) than in patients with COPD, suggesting that neutrophil-mediated chronic inflammation plays a significant role in the pathogenesis of lung cancer [158]. Neutrophils release reactive oxygen species (ROS), which enhance inflammation through the activation of mitogen-activated protein kinases (MAPKs) and redox-sensitive transcription factors, including NF-κB [135]. Activated neutrophils can express serine proteases, including neutrophil elastase (NE), cathepsin G, and proteinase-3, matrix metalloproteinases, such as MMP-8 and MMP-9, as well as other proteins, such as myeoloperoxidase (MPO) and human neutrophil lipocalin, and release them from their granules into the extracellular space. These mediators play important roles in the inflammatory process [131].
Other stromal cells that influence angiogenesis are fibroblasts, which secrete stromal-derived factor 1 (SDF-1) [122], and BM progenitors cells, such as Tie-2-expressing monocytes (TEMs) [99], pericyte progenitors [87, 148], endothelial progenitor cells (EPCs) [52], and hematopoietic progenitor cells (HPCs) [39, 85]. For example, in mouse cancer models, the lungs recruit HPCs, including VEGFR1+ cells and other myelomonocytic cells, leading to microenvironments that favor tumor growth. Particularly striking is the finding that metastatic primary tumors release specific cytokines such as VEGF, SDF-1, TGF-β, and TNF-α, which systemically initiate pre-metastatic niches in distal target organs such as the lungs, characterized by the accumulation of BM-derived cells such as VEGFR1+ HPCs and CD11b+ inflammatory myeloid cells [134]. Furthermore, these pre-metastatic niches are characterized by the induction of chemoattractants such as, S100A8, growth factors, ECM proteins including fibronectin, and ECM-modifying proteins like lysyl oxidase [16, 40, 65, 79], creating a permissive microenvironment for metastasis [134]. Importantly, S100A8/A9 expression in the pre-metastatic niche in turn induces expression of serum amyloid A (SAA) 3, which through the Toll-like receptor 4 (TLR4) leads to the activation of NF-κB signaling and further amplification of inflammatory responses [66]. The pre-metastatic niche has become an exciting area of research in the quest for novel therapeutic and prophylactic strategies against metastasis [149]. In contrast to pre-metastatic niches, a novel mechanism was recently described, whereby metastasis-incompetent tumors generate metastasis-suppressive microenvironments in the lungs by inducing expression of a potent antiangiogenic factor, Thrombospondin 1 (Tsp-1), in the recruited BM-derived myeloid cells [18]. Tsp-1 induction is mediated by the activity of prosaposin (PSAP), a protein secreted by poorly metastatic cells, which acts systemically to reprogram myeloid cells into metastasis-inhibitory cells [18]. COPD and other chronic inflammatory diseases of the lung that may constitute a risk factor for the development of lung cancer are characterized by infiltration of leukocytes into the lung parenchyma. It remains to be determined whether the inflammatory cells in COPD act in ways similar to those in the pre-metastatic niche in lung cancer.
The expression of angiogenic factors by tumor cells or inflammatory cells is a trigger for the formation of new blood vessels. Initiation of angiogenesis relies on an angiogenic “switch” triggered by secretion of angiogenic factors by tumors and stromal cells [10, 45, 51]. Angiogenesis is controlled by pro-angiogenic factors such as bFGF, EGF, MMPs, PlGF, HGF, and PDGF, which have been correlated with poor prognosis in NSCLC [20, 43]. The VEGF–VEGFR signaling pathway is frequently upregulated in lung cancer, and VEGF overexpression is associated with tumor progression [153]. High VEGF levels have been identified as an independent prognostic factor that correlates with poor prognosis in patients with lung cancer [64]. Interestingly, the expression of different VEGF isoforms in human lung cancer is associated with the degree of tumor invasiveness and the patient’s prognosis. In a murine xenograft model of human lung cancer, tumors overexpressing VEGF189 were larger than those overexpressing the other VEGF isoforms [167]. Consistent with this observation, previous studies have shown that VEGF189 mRNA isoform expression ratio showed a greater correlation with tumor angiogenesis, postoperative relapse time, and survival and could be used as a prognostic indicator for patients with non-small cell lung cancers (NSCLC) [123, 167]. Serum VEGF levels are higher in patients with lung cancer than controls [147], while in patients with adenocarcinoma there was a significant correlation of the expression of VEGF165 with tumor size [25]. Overall, patients with higher serum VEGF levels had lower survival compared to patients with lower VEGF levels. The measurement of serum VEGF has also been shown to be a marker of response to chemotherapy, as a decrease of VEGF levels after initiation of chemotherapy correlated with response to therapy [33]. However, given these correlations, not much is known about the molecular mechanisms by which specific isoforms function, and further studies are required to eventually exploit them as potential therapeutic targets.
Moreover, autocrine signaling between bFGF and fibroblast growth factor (FGF) receptor has been shown to be particularly important in NSCLC [105], and circulating levels of bFGF correlated with clinical outcome [14, 33, 64]. Another family of pro-angiogenic factors includes the CXC chemokines containing the glutamic acid-leucine-arginine (ELR) motif, which in humans include CXCL1, CXCL3, CXCL5, and CXCL8 [152]. These chemokines, largely produced by NF-κB transcriptional activation [83], represent an alternative pathway for the angiogenic switch that contributes to angiogenesis, “immunoangiostasis,” and organ-specific metastases in NSCLC [2, 151]. Recently, epidermal growth factor-like domain 7 (EGFL7) has emerged as a mediator of angiogenesis in the lung. EGFL7 is a secreted protein, expressed by ECs, which governs lumen formation during vasculogenesis. Notably, miR-126 harbored within intron 7 of the egfl7 gene plays a crucial role in angiogenesis and maintenance of vessel integrity [119]. Interestingly, a recent study has demonstrated that EGFL7 is a direct target of miR-126 in lung cancer cells, explaining in part the observed effect of miR-126 on tumorigenesis [154]. Given that many key pro-angiogenic factors may trigger and maintain the angiogenic switch, analysis of many NSCLC specimens has revealed that tumors develop unique “angiogenic signatures” characterized by the expression of one dominant angiogenic factor [107].
In NSCLC, the degree of tumor-associated angiogenesis correlates with disease progression and predicts unfavorable survival outcome [34]. In particular, high vascularity at the tumor periphery has been correlated with tumor progression [64]. However, high steady state vessel density in the lung has imposed challenges in accurate identification and quantification of neoangiogenic microvessels in the tumor tissue. Notably, some NSCLCs do not display an angiogenic phenotype and these tumors are invasive, exploiting the preexisting alveolar vessels for growth [70, 120].
Vascular Remodeling in Inflammation and Cancer
In COPD or lung cancer, preexisting vessels proliferate and undergo remodeling. This vascular remodeling involves changes in activity of receptors and adhesion molecules, which leads to the loss of hierarchical organization, and changes in three-dimensional architecture, as well as poor pericyte coverage [7, 110]. The expanded vasculature exhibits increased blood flow, plasma leakage, and influx of inflammatory cells [109]. EC remodeling is associated with increased expression of P-selectin, E-selectin, EphA4, and ICAM-1, typical of sustained inflammation, and is accompanied by the expression of molecules that promote endothelial gap formation and leukocyte rolling, attachment, and migration. Compared to EC phenotypes in inflammation, ECs in tumors undergo disorganized sprouting, proliferation, and regression, and become dependent on VEGF or other factors for survival. The growing vasculature supports tumor growth; however, impaired endothelial barrier function and increased interstitial pressure diminish blood flow and alter immune cell traffic. Despite the rich vessel density, oxygen delivery usually becomes inadequate for tumor cell viability. Inhibition of VEGF signaling in tumors stops sprouting angiogenesis and triggers regression of some tumor vessels while normalizing others [75]. Some normal capillaries in normal thyroid, pancreatic islets, and intestine may also regress after VEGF blockade, but most remodeled vessels at sites of inflammation do not. Pericytes and empty sleeves of vascular basement membrane persist after ECs regress and provide a scaffold for blood vessel regrowth, which can occur within days after VEGF inhibition ends.
The inflammatory response increases vessel permeability and induces endothelial activation, which, when persistent, results in capillary sprouting. Increased vascular permeability contributes to many diseases, including acute respiratory distress syndrome (ARDS), cancer, and inflammation. Critical soluble inflammatory regulators of endothelial barrier function, such as VEGF (originally named vascular permeability factor), TNF-α, TGF-β, and IL-2 have been implicated in the control of vascular permeability. The endothelial lining of the vasculature forms the physical barrier between the blood and underlying tissues. Junctions between adjacent ECs are modulated by vasoactive factors to facilitate the transendothelial migration of leukocytes during inflammation. Leukocytes undergoing diapedesis first attach to ECs’ intercellular adhesion molecule (ICAM-1) through their LFA-1 receptor [21]. Numerous signaling pathways are triggered in both the leukocyte and the EC, and leukocytes start rolling over the endothelial wall. Tumor cells may exploit this pathway in order to colonize organs through extravasation. Activated endothelium is therefore an easy escape route for blood-borne metastatic cells, due to its leakiness and stickiness, which enhances diapedesis-like events by cancer cells. However, the underlying mechanisms of leukocyte-stimulated changes in endothelial barrier function have remained elusive. A full understanding of the signaling cascades that affect endothelial junctions will likely identify novel therapeutic targets to reverse aberrant vascular permeability or enhance leukocyte infiltration into tissues.
Vascular permeability has an important role in cancer biology. Hypoxic conditions in progressing primary tumors result in HIF-1α-mediated induction of VEGF that contributes to vascular permeability. Vascular permeability facilitates intravasation of invasive tumor cells to the circulation, an initial step in the metastatic cascade. Once in the circulation, it is believed that circulating tumor cells may, in many instances, mimic diapedesis in order to extravasate and colonize distant organs. Many tumor cells aberrantly express leukocyte selectins and integrins, allowing them to tether to the endothelial layer to begin the process of diapedesis [97, 116].
Furthermore, it has been demonstrated that TGF-β in the breast tumor microenvironment results in the induction of angiopoietin-like 4 (ANGPTL4) in the cancer cells. ANGPTL4 disrupts vascular EC–EC junctions, increases the permeability of lung capillaries, and facilitates the transendothelial passage of disseminated tumor cells to seed pulmonary metastasis [124]. More recently, it was shown that lung vascular permeability is controlled mechanically by changes in lysyl oxidase-mediated collagen cross-linking and ECM structural changes [104].
Tissue Remodeling in Inflammation and Angiogenesis
An important hallmark that links inflammation and angiogenesis in cancer is tissue remodeling. Multiple proteases are involved in tissue remodeling, of which the neutrophil proteases have key roles. Neutrophil granules carry key proteases, including the serine proteases Cathepsin-G, proteinase 3, and NE, the collagenase MMP-8, and gelatinases like MMP-9 and MMP-2 [56], which are involved in ECM remodeling. Following neutrophil degranulation in the tissue microenvironment, these enzymes released extracellularly play crucial roles in proteolytic processes at sites of inflammation. Neutrophil serine proteases, including NE, can degrade ECM leading to compromised alveolar structures [54]. In addition, ECM degradation releases matrix-trapped factors, including TGF-β and VEGF, increasing their bioavailability. Using the oncogenic K-ras model of lung adenocarcinoma, it was shown that NE-mediated cleavage of insulin receptor substrate 1 (IRS-1) promotes tumor growth via the activation of the PI3K pathway [68]. New generation NE inhibitors are currently being used in early phase clinical trials for COPD and emphysema-like conditions [60, 169]. Other inflammatory cells, such as monocytes and macrophages, easily infiltrate the modified ECM, and by secreting proteases such as the cysteine proteases cathepsins, macrophages further impact the ECM.
Once the inflammation subsides, clearance occurs, whereby macrophages engulf dead cells and debris. Subsequently, fibroblasts infiltrate and secrete collagen and other connective tissue components to restore the ECM [49, 95, 146]. During tissue remodeling, sprouting ECs and tip cells lead their way into the stroma by secreting proteases that degrade the matrix and allow for the invasion of the new sprouting vessels [159]. The fibrotic tissue formed by fibroblasts and the deposited collagen further support the sprouting vessels. Not surprisingly, proteases, including MMPs, which are involved in tissue remodeling during inflammation, also promote angiogenesis. MMP-2 and MMP-9, for instance, release matrix-bound VEGF, increasing its bioavailability [11, 36, 89].
Blockade of MMPs has been considered a potential therapeutic strategy, as this would block angiogenesis, invasion, and infiltration by immune cells. However, MMP inhibition as an anticancer therapeutic strategy has yielded disappointing results, highlighting the need for better insight into the mechanisms by which this growing family of multifunctional enzymes contributes to tumor growth [160].
Paracrine Roles of ECs
ECs that form the vasculature have key functions in providing nutrients and oxygen to the tumor; however, emerging studies have begun to describe “angiocrine” regulation as a major endothelial function in cancer [47]. Vascular ECs actively participate in and regulate the inflammatory response in both normal and diseased tissues [133], and emerging data suggest that ECs directly influence tumor behavior [10, 48]. In a recent study, endothelial-derived angiocrine signals were shown to induce regenerative lung alveolarization. Particularly, activation of VEGFR2 and FGFR1 in pulmonary capillary ECs induced MMP14 expression that unmasked EGF receptor ligands to enhance alveologenesis [31]. Lung ECs also control lung stem cell differentiation, as BMP4-Bmpr1a signaling triggers calcineurin/NFATc1-dependent expression of Tsp-1 in lung ECs to promote alveolar lineage-specific bronchioalveolar stem cell differentiation [93].
Pulmonary vascular endothelial dysfunction is a characteristic pathological occurrence in COPD. As the most important risk factor of COPD, cigarette smoking may initiate pulmonary vascular impairment through direct injury of ECs or release of inflammatory mediators. Morphological changes such as EC damage and EC apoptosis that lead to vessel architecture malformations are highly prevalent in the pulmonary vasculature in COPD patients. Vascular abnormalities impair gas exchange and may result in pulmonary hypertension, which is one of the principal factors associated with reduced survival in COPD patients. Endothelial monocyte-activating protein 2 (EMAP II) is a pro-inflammatory endothelial- and monocyte-activating polypeptide and an antiangiogenic molecule, which specifically induces apoptosis in ECs [55]. EMAP II is upregulated by general cellular stress, hypoxia, and inflammation-promoting bacterial lipopolysaccharide (LPS). EMAP II is also an essential mediator of cigarette smoke-induced lung emphysema in mice, linking EC apoptosis with inflammation, and recently CXCR3 receptor was implicated in EMAP II-induced apoptosis in ECs [57].
miRNA Function in Inflammation and Angiogenesis
miRNAs are emerging as new regulators of inflammation and angiogenesis in carcinogenesis. They are a class of single-stranded, small, noncoding RNAs that typically bind to the 3′ UTR of target mRNA sequences, leading to the reduction of protein expression predominantly by destabilizing target mRNAs and/or by translation inhibition [8]. miR-181b was shown to be a cytokine-responsive miRNA that regulates the expression of key NF-κB-regulated genes involved in the endothelial response to inflammation by regulating the NF-κB signaling pathway [155]. Recently, miR-135b was shown to promote lung cancer by regulating multiple targets in the Hippo pathway. Notably, miR-135 expression is regulated in part by NF-κB signaling, implying the role of inflammation [100]. Similarly, miR-200, by virtue of targeting angiogenesis-promoting cytokines IL-8 and CXCL1, impaired lung cancer in mice [130].
Therapeutic Strategies that Target Angiogenesis and Inflammation
Lung cancer is a global public health problem with an estimated 1.3 million new cases each year [76]. In the USA, approximately 226,160 new cases of lung cancer are diagnosed per year with over 160,000 deaths. Despite advances in treatment options, including minimally invasive surgical resection, stereotactic radiation, and novel chemotherapeutic regimens, the 5-year survival rate in NSCLC remains at ~ 15 %. This is because available targeted therapies such as EGFR tyrosine kinase inhibitors (TKIs, erlotinib, and gefitinib) and EML4-ALK inhibitor (crizotinib) benefit only 15–20 % of NSCLC patients who carry specific drug-sensitive mutations. Even in these patients, acquired resistance is a major impediment to a durable therapeutic response [19, 38, 126]. Therefore, traditional standard cytotoxic chemotherapies remain the only treatment options for the majority of advanced NSCLC patients, and these treatments also usually fail, resulting in an aggressive metastatic relapse. As such, there is an unmet medical need for the development of additional targeted therapies for lung cancer patients.
The relationship between inflammation, innate immunity, and angiogenesis in tumor growth and development has become a focus of recent investigations. Although, many of the molecular and cellular mechanisms mediating this relationship remain unresolved, there has been increased interest in evaluating antiangiogenic, immunomodulatory, and anti-inflammatory agents in the treatment of various malignancies, including NSCLC [12].
The VEGF signaling pathway is central in tumor angiogenesis. Bevacizumab, a humanized monoclonal antibody against VEGF, has been approved in many countries for use in combination with first-line platinum-based chemotherapy (carboplatin and paclitaxel) for the treatment of NSCLC patients with advanced stage disease [63, 132]). Approvals were based upon an improvement in response rate (RR) and progression-free survival (PFS) seen with the addition of bevacizumab to chemotherapy in two large phase III studies, the North American Eastern Cooperative Oncology Group (ECOG) 4599 [142] and the European AVAiL [136]. However, not all trials showed significant improvement in overall survival (OS). Despite the favorable results with bevacizumab, trials with other humanized antibodies targeting VEGF receptors have failed to meet their primary end points. Aflibercept (VEGF Trap), an FDA-approved drug for metastatic colorectal cancer, has been evaluated in second-line therapy of NSCLC. Aflibercept is a recombinant VEGF receptor–antibody protein fusion with higher affinity for VEGF-A than bevacizumab, plus affinity for VEGF-B and placental growth factor (PlGF). By attaching itself to these ligands, aflibercept acts as a decoy receptor for VEGF, preventing cognate pro-angiogenic ligands from binding to receptors on blood vessel cells and stimulating angiogenesis [71]. A randomized phase III trial of second-line docetaxel with or without aflibercept in platinum-pretreated patients with advanced nonsquamous NSCLC failed its primary end point of overall survival despite higher response rates and progression free survival in the experimental arm [118]. Ramucirumab, a humanized monoclonal antibody targeting the extracellular domain of VEGFR2, has also been evaluated in a phase III randomized trial of second-line docetaxel with or without ramucirumab. The trial did not meet its primary end point.
The encouraging results with bevacizumab have led to the development of other antiangiogenic agents, including small molecule TKIs targeting the VEGF receptor (VEGFR). Motesanib, a selective oral inhibitor of VEGF receptors-1, 2, and 3, platelet-derived growth factor receptor (PDGFR), and c-Kit was tested in a randomized phase II trial in combination with carboplatin/paclitaxel as frontline therapy for patients with advanced NSCLC, and results showed that RR, PFS, and OS were comparable in those patients receiving either motesanib or bevacizumab [13]. However, an international randomized phase III trial with carboplatin/paclitaxel either alone or in combination with motesanib in patients with advanced NSCLC showed no improvement in overall survival compared with placebo; despite an improvement in PFS and overall response [28, 143]. Another phase III trial evaluated the addition of the multi-kinase inhibitor (including VEGFR2) sorafenib to chemotherapy in patients with advanced nonsquamous NSCLC. Again, despite a slight but statistically significant improvement in PFS, there was no improvement in OS, the trial’s primary end point [129]. Similarly, a randomized trial of vandetanib, an EGFR/VEGFR/RET kinase inhibitor, did not demonstrate improvement in OS compared to placebo in the second-line setting. A phase II/III trial of cediranib, a small molecule inhibitor of VEGFR and PDGFR, did not proceed to phase III due to the high toxicity associated with cediranib at the dose selected for the trial [92]. A recently reported phase III trial assigned patients with advanced NSCLC who failed first-line therapy to docetaxel with and without nintedanib, a multi-angiogenic kinase inhibitor (VEGFR1-3/FGFR1-3/PDGFR/FLT3). Nintedanib in combination with docetaxel was associated with significant improvement in PFS and OS especially in patients with adenocarcinomas [137]. This is the first and only trial to demonstrate an improvement in OS using a targeted agent in the second-line setting. Finally, a phase III placebo-controlled trial of carboplatin and paclitaxel with and without the vascular disrupting agent vadimezan (ASA404) as first-line therapy for patients with advanced lung cancer did not meet the specified primary and secondary end points of OS and PFS [88, 111, 112].
Ongoing clinical trials involving several new antiangiogenic therapies, including cediranib, sunitinib, pazopanib, brivanib, ABT-869, axitinib, ABT-751, and NPI-2358, are underway and several of these agents have shown promising phase I/II results. Results from recently completed and ongoing phase III trials will determine if these newer antiangiogenic agents will be incorporated into clinical practice [1].
However, in aggregate, antiangiogenic therapy has not yet had the impact some have hoped for. Many clinical benefits are short-lived; while numerous trials have shown an increase in survival of patients treated with antiangiogenic therapy, the increase for many was a matter of months [82]. Several possibilities have been suggested to explain why VEGFR TKIs trials have not yielded significant benefit in NSCLC. For example, lack of predictive biomarkers continues to be a major hurdle in the selection of adequate patient cohorts that are most likely to benefit. Some studies have alluded to a possible link between antiangiogenic therapy and increased metastasis in multiple tumor types [35, 125].
Compared to advances with antiangiogenic therapies, success with anti-inflammatory treatments has been less impactful. Previous clinical trials have indicated that long-term use of aspirin or other NSAIDs decreases the incidence of colorectal, esophageal, breast, lung, and bladder cancers [162]. While initial studies had focused on various broad-spectrum NSAIDs (which nonspecifically inhibit both COX-1 and COX-2), more recent studies have examined COX-2-specific agents, such as celecoxib [90]. Significant preclinical and clinical data support the importance of COX-2 in development and progression of NSCLC. Despite this, a protective effect of NSAIDs was not observed on lung cancer development in either the general or high-risk COPD populations [161]. Moreover, clinical trials of COX-2 inhibition in NSCLC have been disappointing [108]. The lack of clinical benefit in the Cancer and Leukemia Group B (CALGB) 30203 trial suggests that COX-2 inhibition would be of value in COX-2-overexpressing tumors, emphasizing the need for a prospective, randomized trial that selects patients for therapy on the basis of COX-2 expression [36]. CALGB 30801 is a randomized phase III double-blind trial evaluating selective COX-2 inhibition in COX-2 expressing advanced NSCLC. However, given the gastrointestinal (GI) toxicity and nonspecific activity of NSAIDs, and the cardiotoxicity of specific COX-2 inhibitors, the use of such agents continues to remain controversial [32].
Two recent studies have shed light on the future therapeutic potential of the NF-κB-mediated inflammatory pathway in lung cancer. Logsdon and colleagues found that in the presence of oncogenic Ras, inflammatory stimuli initiate a positive feedback loop involving NF-κB that further amplifies Ras activity to pathological levels [27]. Because a large proportion of lung cancer patients possess Ras mutations, disruption of this positive feedback loop may be an important strategy for cancer prevention. In another study, using mouse models of lung cancer, Verma and colleagues found that therapies targeting the enzyme IKK2 (involved in inflammation) and Timp1, which helps activate the body’s inflammatory response, may effectively treat certain lung cancers [165].
Major efforts are required to unravel crosstalk signaling pathways between inflammatory and angiogenic components in NSCLC. This is a major challenge given the high heterogeneity of genetic and epigenetic alterations present in different cancers, differences in host genetic background, as well as tissue-specific responses. Understanding the cellular and molecular mechanisms underlying these processes will provide novel avenues leading to the discovery of biomarkers for disease stratification, molecular diagnosis and prognosis, and devising therapeutic strategies against lung cancer
Acknowledgments
This project was partly supported by the Cornell Center on the Microenvironment and Metastasis through Award Number U54CA143876 from the NCI to VM. RC was supported by the National Lung Cancer Partnership (NLCP), and fellowships from the “Government of Navarra” and the “Camara Navarra de Comercio” (Navarra, Spain).
References
1.
Aggarwal C, Somaiah N, Simon G (2012) Antiangiogenic agents in the management of non-small cell lung cancer: where do we stand now and where are we headed? Cancer Biol Ther 13:247–263PubMedCentralPubMed
Related posts:
 Inflammation and Lung Cancer: Molecular Pathology
Inflammation and Lung Cancer: The Role of Epithelial–Mesenchymal Transition
Inflammation and Lung Cancer: Addressing Inflammation with Immunotherapy
Inflammation and Lung Cancer: Prevention
Inflammation and Lung Cancer: Molecular Pathology
Inflammation and Lung Cancer: The Role of Epithelial–Mesenchymal Transition
Inflammation and Lung Cancer: Addressing Inflammation with Immunotherapy
Inflammation and Lung Cancer: Prevention
 Inflammation and Lung Cancer: Eicosanoids
Inflammation and Lung Cancer: Eicosanoids
 Inflammation and Lung Cancer: The Relationship to Chronic Obstructive Pulmonary Disease
Inflammation and Lung Cancer: The Relationship to Chronic Obstructive Pulmonary Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


