Fig. 6.1
Pathways controlling eicosanoid production. Arachidonic acid is incorporated into membrane glycerophospholipids. Activation of cPLA2 hydrolyzes these lipids to produce free AA. Alternatively, AA can be derived from 2-arachidonylglycrol through the action of monoacylglycerol lipase (MAGL). Arachidonic acid (AA) converted through AA is metabolized through the COX pathway to produce prostaglandins prostacyclin and thromboxane. The 5-lipoxygenase pathway requires 5-lipoxygenase activating protein (FLAP) to produce 5-HpETE which can be converted to 5-HETE through a specific reductase. Alternatively, 5HpETE is converted to LTA4. This in turn is converted by LTA4 hydrolase to LTB4. During transcellular biosynthesis, LTA4 can be secreted and taken up by another cell expressing LTA4 hydrolase, and converted to LTB4. LTC4 synthase converts LTA4 to LTC4, which is subsequently converted to LTD4 by γ-glutamyl transpeptidases, and then to LTE4 by dipeptidases. 12/15 lipoxygenase produce 12-HETE and 15-HETE from AA. Finally, cytochrome P450 members, particularly CYP2J2 and CYP2C8 produce a family of EETs as well as 20-HETE
Finally, the cytochrome P450 epoxygenases, specifically CYP2J2 and CYP2C8, produce epoxygenated fatty acids (EETs), including 5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET. These enzymes also posseses ω-hydroxylase activity [46], which results in production of distinct HETEs, including 9-, 11-, 16-, 17-, 18-, 19-, and 20-HETE. Over 100 distinct eicosanoid species have been identified [20]. An additional level of complexity is derived from the fact that distinct products may be produced through multiple pathways. For example, HETEs can be derived from all three pathways; even COX-1 has been shown to produce 15(S) HETE in the setting of inhibition by aspirin [47]. In general, the capacity of a particular cell type to produce a specific eicosanoid will depend on the repertoire of enzymes that are expressed, as well as the presence of factors that activate the pathway. For example, production of PGI2 will require activation of cPLA2, the expression of COX-1 or COX-2, as well as expression of prostacyclin synthase (PGIS), whereas production of LTC4 will in general require expression of 5-lipoyxgenase as well as LTC4 synthase. An exception to this paradigm has recently been described, which adds another level of complexity. Transfer of biosynthetic intermediates in the eicosanoid pathway has been shown to occur [48]. In this process, which has been termed transcellular biosynthesis, PGH2, an intermediate in the COX pathway, and LTA4, an intermediate in 5-lipoxygenase pathway, have been shown to be transferred from one cell to another. Thus, coordination between two cell types, each of which lacks the full complement of enzymes to produce a specific eicosanoid, results in production of novel products .
The majority of eicosanoids are secreted from cells and act in an autocrine or paracrine fashion through a family of G-protein coupled receptors [10). For some eicosanoids, multiple receptors have been identified. For example, PGE2 can signal through four receptors, designated as EP1–4 [49, 50]. These receptors couple to distinct effector pathways, with EP2 and EP4 signaling through increases in cAMP, whereas EP3 inhibits cAMP; EP1 signals through phospholipase C, and increases in intracellular Ca2+ [10]. For other eicosanoids, such as PGI2, only a single receptor has been identified. Multiple leukotriene receptors have been identified. Two receptors specific for LTB4 have been identified, a high affinity receptor designated BLT-1 [51], and a low affinity receptor designated BLT-2 [52]. BLT-1 appears to signal through Gq, whereas BLT-2 appears capable of signaling through both Gq and Gi. In addition to two cysteinyl leukotriene receptors, designated as CysLT1 and CysLT2, have been identified [40]. These receptors show different patterns of cell expression and can bind LTC4, LTD4, and LTE4, with different affinities . They both are coupled to Gq, and signal through increases in intracellular Ca2+ .
Studies in a variety of cancer have implicated individual eicosanoids as mediators of cancer initiation, as well as progression and metastasis [53]. Most extensively studied are prostaglandins , specifically PGE2, produced through the action of COX-1/2, and microsomal PGE2 synthase (mPGES). However, more recent work has implicated other eicosanoids including products produced through both the lipoxygenase and cytochrome P450 pathways. In this review, we will examine the complexity of these products in regulating lung cancer initiation as well as progression and metastasis. While the potential of different cell types within a tumor to produce distinct spectra of eicosanoids underscores the complexity of the field, the development of a large number of preclinical models and specific agonists and antagonists will provide keys to a better understanding of the role of these molecules. In addition, these compounds represent attractive chemotherapeutic and chemopreventive agents for cancer.
Control of Eicosanoid Production
Eicosanoids are produced in a cell-specific and time-dependent fashion. Since these molecules are generally short-lived, and act in an autocrine or paracrine fashion, it is critical that their production be under dynamic regulation. As might be anticipated, this is mediated through a number of regulatory controls, including transcriptional, posttranscriptional, and posttranslational pathways. In general, individual cell types will have the capacity to produce a restricted spectrum of eicosanoid products. This will, in general, be determined by the expression pattern of enzymes required for production, processing, and potentially, cell export. For example, production of PGE2 requires that cells express a PLA2 isoform, most commonly cPLA2-α, a cyclooxygenase isoform (COX-1 or COX-2), as well as the downstream specific synthase, in this case PGE synthase, of which there are three isoforms [54, 55]. Expression of these enzymes can be controlled by exogenous stimuli, Whereas COX-1 is not usually dynamically regulated, COX-2 was initially identified as an immediate early gene, and is rapidly induced in many cell types in response to growth factor or inflammatory cytokine stimulation [56]. While regulation of cPLA2 expression can also be mediated through growth factor or oncogene signaling, acute regulation of this enzyme is mediated through increases in intracellular Ca2+ concentration, as well as regulatory phosphorylation through ERK signaling [57, 58]. Upon elevations in intracellular Ca2+ , cPLA2 translocates in most cells to the nuclear envelope/endoplasmic reticulum [23, 59], which is in close physical proximity with downstream enzymes, including COX and 5-lipoxygenase. It is presumed that interactions between cPLA2 and downstream enzymes are mediated through specific protein–protein interactions. Specific cPLA2 binding proteins have been identified [60], but their role in eicosanoid production has not been established.
In addition, PGE2 production will be subject to negative regulation by enzymes that metabolize PGE2, specifically 15-hydroxy-PGE2 dehydrogenase (15-PGDH). In addition to increased expression of COX-2, lung tumors show decreased expression of 15-PGDH, which will further augment steady-state levels of PGE2 [61–63]. This enzyme is regulated transcriptionally, and potentially posttranscriptionally [64] by thiazolidinediones, which are know activators of the nuclear receptor PPARγ. This illustrates that single agents may regulate PGE2 production through multiple pathways. Activation of PPARγ can simultaneously directly inhibit expression of COX-2 via NF-κB, and increase expression of 15-PGDH, which will synergize to strongly block PGE2 production [65–67].
5-lipoxygenase is also regulated through a number of pathways [68, 69]. 5-lipoxygenase is expressed primarily in leukocytes, including neutrophils, eosinophils, and macrophages , as well as B-lymphocytes. Normally, expression levels of 5-lipoxygenase are low in epithelial cells or cancer cells; however, expression has been detected in some human non-small-cell lung cancer (NSCLC) in response to cytokine stimulation [70]. 5-lipoxygenase, similar to cPLA2 contains a C2 domain, which is a Ca2+ -phospholipid binding domain [68], and in most cell types increases in intracellular Ca2+ are required for enzymatic activity. Increased Ca2+ causes translocation of the enzyme to the nuclear envelope where it is in close proximity with 5-LO activating protein (FLAP) [71]. FLAP is an integral membrane protein involved in presentation of AA to the 5-lipoxygenase, and is the target for the drug MK-886. 5-lipoxygenase can also be localized in part to the nucleus, and the enzyme contains a nuclear import sequence [72] In addition, 5-lipoxygenase has been shown to be regulated by protein phosphorylation [73]. Phosphorylation appears to mediate export out of the nucleus, which may result in increased production of 5-lipoxygenase products [74]. The importance of these phosphorylation events has not been fully defined. Activation of 12/15-lipoxygenase also requires increases in intracellular Ca2+, which mediates translocation to a membrane compartment [75]; however, other signaling mechanisms, which may regulate enzymatic activity in inflammatory cells, have not been characterized.
Cancer Development
A large body of work over the past 30 years has defined the oncogenic pathways that transform “normal” epithelial cells resulting in cancer cells [76, 77]. Over the past 5 years, it has become clear in several types of cancers, and prominently in lung cancer, that different oncogenic drivers result in distinct classes of cancer, which in fact represent distinct diseases. NSCLC, which comprises approximately 80 % of lung cancer, can be subdivided into distinct subgroups based on oncogenic drivers, and more recently similar classifications have been applied to squamous lung cancer [78, 79]. These areas have been extensively reviewed, and will not be discussed in detail here. However, in addition to sensitivity to specific therapeutic agents, different oncogenic drivers will have differential effects on production of eicosanoids . Mutations in oncogenic K-Ras have been observed in approximately 30 % of the adenocarcinomas. A large fraction of NSCLC not expressing K-Ras either have somatic mutations in receptor tyrosine kinases epidermal growth factor receptor (EGFR), fusions with other receptors (e.g., AML-ELK4), or are driven through receptors which are not mutated, but may be overexpressed (FGFR) [80–82].
Over the past 10 years, it has become evident that cancer progression and specifically metastasis require complex interactions between the transformed epithelial (tumor) cell and the surrounding tissue, termed as the tumor microenvironment (TME)) [83–91]. This complex stroma comprises vascular cells, fibroblasts, extracellular matrix, and both innate and adaptive immune cells. Elegant studies have demonstrated that these cells are critical for cancer progression, and a model has been proposed in which cancer cells promote alterations in the phenotype of stromal cells which result in cells which contribute actively to tumor progression [85, 92]. In response to injury or other type of insult, the immune system response is initially characterized by production of TH1 cytokines such as IL-12, interferon-γ, and TNF-α [93]. These molecules are associated with an antitumorigenic response. As injury resolves, the phenotype of the immune response changes to what is designated as TH2. This is associated with production of pro-angiogenic and antiapoptotic cytokines such as IL-4, IL-13, IL-6, and IL-10. The production of these molecules is usually involved in wound healing. As the insult is removed the immune system reverts to an unstimulated state. However, in the setting of a chronic insult, such as the presence of a tumor, the inflammation does not resolve.
For example, studies examining macrophages in lung cancer have defined polarized phenotypes of these cells, which have been designated as M1 or M2, analogous to the characterization of T cells and TH1 or TH2 [94]. Circulating monocytes are recruited to the site of tumor and are polarized to a classically activated inflammatory macrophage, which presumably acts to inhibit tumor growth. However, through reciprocal interactions between tumor cells and macrophages, as well as potentially other cell types, these macrophages undergo conversion to an alternatively activated phenotype which promotes tumor progression, at least in part through production of growth factors and inflammatory cytokines . In fact, examination of human lung tumors has correlated inflammatory macrophages of the M1 phenotype in tumor islets with extended survival [95, 96]. In contrast, increased M2 macrophages have been associated with a poorer prognosis [97].
While this classification may be useful, it is clear that phenotypes of macrophages represent a continuum of states, and that the emphasis needs to be on the function of these cells, rather than on using a limited set of markers. Similar phenotypes have also been defined for neutrophils, which can be classified as either N1 or N2, analogous to the macrophage designations [98]. Macrophages and other inflammatory cells such as neutrophils are potent producers of eicosanoids, most prominently through the lipoxygenase pathways. The role of these eicosanoids in promoting cancer progression will be discussed below.
Lung Cancer and Inflammation
The link between inflammation, specifically chronic inflammation and development of cancer has been described for a long time [99]. Environments in which there is a chronic inflammatory response are linked to increased incidence of cancer. Alternatively, the presence of a tumor results in the mounting of an inflammatory response, which, in itself, can promote progression and metastasis of the tumor. Specifically in the case of lung cancer, exposure to cigarette smoke, or other environmental toxins mediates increased incidence of chronic obstructive pulmonary disease (COPD), which is associated with an increased risk for lung cancer [100, 101]. The majority of lung cancer cases arise in either current or former smokers. Chronic exposure to tobacco smoke promotes oxidative stress and induces a widespread inflammatory infiltrate [102]. This is comprised of increased numbers of macrophages, dendritic cells, granulocytes, and activated lymphocytes [103]. It is assumed that these infiltrating cells result in a microenvironment that promotes lung cancer initiation and progression. Elevations in eicosanoid production have been reported in inflammatory lung diseases such as COPD [104] and airway inflammation [105]. The relationship between these products and the increased incidence of lung cancer associated with these diseases has not been well examined. However, targeting leukotriene production has been proposed as a potential therapeutic for treatment of COPD [106].
Eicosanoids in Lung Cancer
Lung cancer has classically been divided into small cell lung cancer (SCLC) and NSCLC [107]. Eicosanoids have been studied extensively in NSCLC, and the role of these products in SCLC is less developed [108]. In general, the eicosanoid profile of human lung cancer cell lines is limited to products of the cyclooxygenase pathway. Increases in prostaglandin production have been demonstrated in a subset of NSCLC [109–111]. Elevated expression of cPLA2 and COX-2 resulting in high PGE2 has been associated with NSCLC cell lines expressing oncogenic K-Ras [112). Inhibition of prostaglandin production via blocking either cPLA2 or COX-2 inhibits transformed growth of NSCLC, and the development of tumors in mice in response to chemical carcinogens [35, 113, 114]. In human tumors, early studies using mass spectrometry approaches defined changes in multiple COX products in human lung tumors, compared to uninvolved tissue from the same patient [109].
Using pharmacological and molecular approaches to inhibit this pathway, it has been shown that PGE2 contributes to cancer progression at several stages. A few other studies have focused on the role of lipoxygenase products, and these have also been implicated as mediating cancer initiation/progression [115–117]. In contrast, PGI2, which is also produced downstream from COX enzymes, has been shown to inhibit lung cancer initiation, as well as having antimetastatic effects. In vivo, it is likely that complex changes in multiple eicosanoids occur during cancer progression .
In the majority of these studies, little is known regarding the specific mechanisms whereby individual eicosanoids regulate lung cancer initiation or progression, and the use of inhibitors does not distinguish between eicosanoids produced by different cell types. To date, there has not been to our knowledge studies to systematically examine a broad spectrum of eicosanoids in the setting of cancer. The advent of mass spectrometric approaches has allowed the measurement of multiple eicosanoid products from both cells and tissues [118]. This technique allows an unbiased quantitative determination of a large group of (> 25) distinct products. In this review we will examine the evidence for distinct eicosanoids in various stages of lung cancer initiation, progression, and metastasis. However, the complexity of this pathway is highlighted by different cells producing distinct profiles of eicosanoids, and a dynamic crosstalk between the distinct pathways. The availability of pharmacological inhibitors and activators of various enzymes in the pathway make this an attractive therapeutic target. Developing a better understanding of the dynamic nature of eicosanoid production will be critical in designing therapeutic strategies for inhibition of lung cancer .
Role of cPLA2 in Lung Cancer
Since cPLA2 represents the rate limiting step in eicosanoid biosynthesis, the role of this enzyme on development and progression of lung cancer has been examined. In mice that are globally deficient in cPLA2 [119], a decrease in lung tumor size was observed in response to administration of urethane, a well-characterized chemical carcinogenesis model [35]. This was also associated with a decreased inflammatory response [34]. In these studies, tumorigenesis was associated with increased levels of PGE2 in the lung, and this was blunted in the cPLA2 knockout mice. More recent studies have examined the role of cPLA2 in lung cancer progression and metastasis. These studies employed an immunocompetent orthotopic model, in which murine lung cancer cells are directly injected into the lungs of syngeneic mice. When identical tumor cells were injected into wild-type or cPLA2 knockout mice, there was no significant effect on primary tumor size, but there was a marked impairment in formation of secondary tumors and distant organ metastasis [120]. Employing bone marrow transplants, it was shown that effects of cPLA2 deletion in the microenvironment are mediated at least in part through bone marrow-derived cells, most likely macrophages. Further studies have focused on the role of cPLA2 on tumor angiogenesis [121–123]. Activation of cPLA2 has been shown to stimulate endothelial cell proliferation and formation of a functional tumor vascular network [122]. Using a pharmalogical cPLA2 inhibitor, it was shown that inhibiting cPLA2 blocked tubular formation in irradiated endothelial cells. In addition, treatment with the inhibitor decreased tumor vascularity and tumor blood flow, resulting in slower tumor growth. Lung cancer cells grown subcutaneously in cPLA2 knockout mice showed slower growth and an attenuated tumor vasculature [121]. Interestingly, this study showed that the defect in endothelial cell proliferation seen with loss of cPLA2 could be reversed by the addition of exogenous lysophospholipids. The vast majority of studies have focused on the role of cPLA2 in arachidonic acid release and subsequent production of eicosanoids . Since cPLA2 is critical for eicosanoid production [23], studies using global cPLA2 knockout mice will not be able to address the role of this enzyme in specific cell type and define the critical downstream eicosanoids. In addition, the lysophospholipids produced by activation of cPLA2 may also play a role in lung cancer. In fact, lysophosphatidylcholine, which is a product of cPLA2 activity has been shown to inhibit metastasis of melanoma cells to the lung [124]. Additional studies examining the effects of specific lysophospholipids in lung cancer are required to define the potential of this other arm of cPLA2 activity.
Finally, cPLA2 belongs to a large family of enzymes. The role of other forms of PLA2 in cancer has not been examined in detail. Several studies have demonstrated that one of the secretory forms of PLA2, sPLA2 IIA may play a role in esophageal cancer [125, 126], and more recently in lung cancer [127–129]. The relationship between the regulation of this enzyme and eicosanoid production remains to be established.
Role of Cyclooxygenase Products
PGE2
PGE2 is the most abundant eicosanoid observed in the setting of lung cancer initiation, progression, and metastasis, and has been the most studied. The PGE2 is constitutively produced at high levels by a significant subset of NSCLC [130]. This is associated with increased expression of both COX-2 as well as mPGES [55]. Blocking production of PGE2 or inhibiting binding to PGE2 receptors blocks proliferation [112, 131], indicating autocrine effects in these cells. While the precise signaling pathways driving these effects have not been completely defined, it has been shown in colon cancer cells that PGE2 can lead to transactivation of the EGF receptor pathway [132].
In addition to promoting growth tumor cells, PGE2 produced by these cells acts on the TME to promote cancer progression. Several studies have shown that PGE2 leads to suppression of dendritic cell antigen presenting activity [133, 134]. In addition, PGE2 can promote tumor-induced immunosuppression by increasing the number or promoting activation of regulatory T cells (Treg) through induction of FOXP3 expression [135], and myeloid derived suppressor cells (MDSC) [136]. It has been reported that crosstalk between cancer cells and MDSC results in upregulation of COX-2 and mPGES in the MDSC, which contributes to the immunosuppressive activity [137]. Production of PGE2 by human NSCLC is critical for suppression of IL-12 and induction of IL-10 in lymphocytes and macrophages [138]. This would be expected to promote an alternatively activated M2 phenotype in macrophages, which results in promotion of cancer through a variety of mechanisms including production of pro-angiogenic growth factors such as VEGF, and IL-6 [120, 139]. PGE2 produced by cells of the TME is important for cancer progression and angiogenesis . Deletion of mPGES-1 in bone marrow-derived cells impairs tumor angiogenesis and enhance tumor growth [140].
The role of specific receptors in mediating the effects of PGE2 has been examined. Deletion of the EP-2 receptor protects mice against the development of lung tumors in a chemical carcinogenesis setting [141]. On the other hand, using a model in which tumor cells are implanted into the lung, it was shown that EP3 knockout mice have slower tumor growth with decreased angiogenesis [142], implicating this receptor. Pharmacologic antagonists directed against the EP4 receptor inhibit lung carcinoma cell migration and invasion [143]. Using a model in which Lewis Lung carcinoma cells are implanted in the lung, it was shown that PGE2 produced by tumor cells signals through EP3 and EP4 in fibroblasts to modify the SDF-1/CXCL4 axis, implicating these receptors on stromal cells [144]. Thus, it appears that signaling through EP2, EP4, and possibly EP3 mediate lung cancer progression. The data regarding the role of EP1 is less clear. In breast cancer, activation of EP-1 suppresses breast cancer metastasis [145]. However, other studies in lung cancer have shown that activation of EP-1 in combination with Her-2/Neu increases production of VEGF-C and promotes lymphangiogensis [146]. In many of these studies, the critical cell type in which these receptors are acting have not been identified. Thus, different PGE2 receptors on distinct cell populations have multiple effects in lung cancer, underscoring the complexity of the eicosanoid pathways. Finally, other factors produced by tumor cells can mediate increased production of PGE2 in cells of the microenvironment. For example, it has been reported that VEGF produced by lung tumor cells leads to induction of PGE2 production by endothelial cells, which in turn suppresses T-cell function [147]. Recently, it has been shown that COX-2 expression is critical for macrophage migration in response to chemoattractants [148]. It is thus important to view PGE2 and other eicosanoids as part of a complex communication between different cell types.
Prostacyclin (PGI2)
PGI2 produced downstream of COX enzymes, through the action of PGIS. In non-tumor bearing lung, PGIS is expressed most highly in endothelial cells, where it regulates vascular tone [149, 150]. Studies using mice with targeted overexpression of PGIS to distal epithelial cells of the lung showed impairment in lung tumor formation, both in response to chemical carcinogens as well as exposure to cigarette smoke [151, 152]. While the mechanism of these protective effects is not completely understood, the effects of PGI2 appear to be independent of the cognate PGI2 cell surface receptor (IP), but rather involve activation of nuclear receptors of the PPAR family [153]. The role of inflammatory cells in mediating these effects remains to be determined. Interestingly, in the setting of tumor inhibition with the PGI2 analog iloprost, increased numbers of macrophages were detected in the lung [153]. The role of eicosanoids in inflammatory cells in the setting of chemoprevention remains poorly understood, and may provide novel chemopreventive agents. However, a recent clinical trial using the orally active PGI2 analog iloprost showed an improvement in the degree of dysplasia in ex-smokers [154]. This is one of the few positive chemoprevention trials in lung cancer. The role of prostacyclin in lung cancer progression has not been well-defined. Early studies have shown antimetastatic effects of PGI2 in an experimental model of metastasis [8]. However, these have not been followed up.
PGD2
Less work has been performed regarding the role of PGD2. PGD2 is produced by inflammatory cells, particularly mast cells, and contributes to inflammation in the lung [155, 156]. Increased levels have also been detected in human lung tumors [109, 157]. Studies using a targeted deletion of the PGD2 receptor (DP) showed increased angiogenesis in an implanted lung tumor model, suggesting that PGD2 is a suppressor of angiogenesis [37]. PGD2 can be metabolized through a nonenzymatic pathway to produce 15-deoxy-∆12,14 prostaglandin J2 (15-PGJ2), which has been shown to be a potent activator of the nuclear receptor PPARγ [158]. Activation of PPARγ by this ligand can induce apoptosis in NSCLC and promote increased production of reactive oxygen species (ROS) [159, 160]. It is thus possible that the anti-tumorigenic effects of PGD2 may be mediated through both receptor-dependent and receptor-independent pathways. However, whether the production of 15-PGJ2 is regulated and actually produced in physiologic amounts in vivo remains controversial [161].
Thromboxane
Increased production of thromboxane A2 had been detected in human lung tumors [162]. Thromboxane can act directly on lung cancer cells to promote proliferation [163] as well as inhibit apoptosis [164]. In addition, this eicosanoid can modulate the microenvironment. Activation of the thromboxane receptor (TP) in NSCLC cells increases expression of monocyte chemoattractant protein-1 (MCP-1) and recruits macrophages to promote invasion of lung cancer cells [165]. In addition, it can regulate production of VEGF, thereby influencing tumor angiogenesis [166]. Mice with targeted deletion of the thromboxane receptor (TP) have been shown to have impaired recruitment of bone marrow-derived cells to the lung in a B16 melanoma colonization model [166]. In addition, mice with deletion of thromboxane synthesis have been developed, but not examined in the setting of any cancer [167]. A pharmacological inhibitor of thromboxane synthase has been developed and is currently in a number of clinical trials for ischemic stroke [168]. Additional investigation using both genetic mouse models as well as pharmacological agents in preclinical lung cancer models is needed to validate the observations obtained in vitro.
Role of Lipoxygenase Products
5-lipoxygenase
As discussed above, 5-lipoxygenase can produce both HETEs and leukotrienes . While certain tumor cells can produce leukotrienes, the major producers of this class of eicosanoids are inflammatory cells—specifically monocyte/macrophages and neutrophils. Pharmacological inhibitors of 5-lipoxygenase such as Zileuton have been shown to inhibit lung tumor formation using a chemical carcinogenesis model [169, 170). However, many of the 5-lipoxygenase inhibitors are not specific and have many off-target effects, which are described in [171]. In general, little is known regarding the role of specific products of 5-lipoxygenase and their impact on either lung cancer initiation or progression.
Earlier studies demonstrated that 5-lipoxygenase products produced by peritoneal macrophages had tumoricidal activity, although the specific product was not identified [172]. Although studies in colon cancer using 5-lipoxygenase knockout mice have demonstrated increased tumor formation and macrophage recruitment [173, 174], similar studies have not been performed in lung cancer preclinical models. Genetic deletion of 5-lipoxygenase in bone marrow-derived cells resulted in decreased production of IL-12 and increased production of IL-10, characteristic of an alternatively activated macrophage [175]. This would suggest that potentially 5-lipoxygenase may have inhibitory effects on tumor progression through modulation of macrophage phenotype. More recent studies have implicated LTB4 in neutrophil recruitment. LTB4 is present in pleural effusions and is associated with increased neutrophil recruitment [176]. However, the source of the LTB4 has not been determined, and the role in tumor progression remains to be established.
HETEs promote increased proliferation in many cell types. They have been shown to mediate the proliferative effects of growth factors such as EGF [177, 178], as well as being mitogenic as sole agents [179]. On the other hand, lipoxins, generated by 15-lipoxygenase in the setting of aspirin treatment, inhibit the growth of lung cancer cells [180].
12/15-Lipoxygenases
The role of these enzymes has not been extensively examined in any cance r. Expression of 15-LOX-1,2 has been shown to be reduced in an NNK model of chemical carcinogenesis [181]. Loss of 15-lipoxygenase expression has also been shown to occur in human lung cancer tissue compared with non-tumor control [181], suggesting that this gene may function as a tumor suppressor. Loss of 15-lipoxygenase is associated with lower levels of 15(S)-HETE and 13(S) HODE. These eicosanoids have been shown to be endogenous activators of PPARγ. Thus lower levels of these eicosanoids would result in decreased activity of PPARγ, which is observed in NSCLC. Exogenous 15-HETE has been shown to inhibit the growth of A549 cells, a human adenocarcinoma cell line [75], also implicating this pathway as a potential tumor suppressor. Earlier studies have defined a role for 12-HETE in mediating regulation of protease activity by cancer cells [182], but the importance of this in vivo has not been examined.
Role of Cytochrome P450 Products
There has been less work regarding products of the cytochrome P450 pathway. However, recent studies have demonstrated that EETs produced by this pathway may play a critical role in metastasis. Cytochrome P450 enzymes have two distinct activities: ω-hydroxylase activity to produce HETEs, principally 20-HETE, and epoxygenase activity to produce EETs [46]. These molecules have been implicated in vascular biology and angiogenesis. However, recent work has demonstrated a role for EETs in tumor growth, promotion of metastasis, and escape from dormancy [183]. In these studies, increased levels of EETs in endothelial cells, through targeted overexpression of either CYP2C8 or CYP2J2, resulted in increased tumor growth in multiple syngeneic tumor models, including growth of Lewis lung carcinoma cells, as well as increased distant organ metastases. Interestingly, the effects on metastasis do not appear to involve the primary tumor, but instead regulate interactions between tumor cells and endothelial cells at the site of the distant organ metastasis. Consistent with these data, addition of a stable 20-HETE analog or overexpression of CYP4A11 was associated with the increase in 20-HETE production and increased invasiveness and expression of VEGF in NSCLC. Treatment of A549 cells with HET0016, a CYP ω-hydroxylase inhibitor, or a 20-HETE antagonist (WIT002) inhibited invasion with reduction in VEGF and MMP-9. [184).
These studies have important implications clinically, since EETs are believed to have beneficial effects on the vasculature, and inhibitors of the enzyme that degrade these products, such as soluble epoxide hydrolase, are in clinical trials in vascular diseases [185). A limited number of studies have examined effects of cytochrome P450 on cancer cell proliferation. Administration of eicosapentaenoic acid (EPA) inhibits proliferation of a limited number of human lung cancer cells [186].
Therapeutic Approaches
As mentioned above, modulators of the eicosanoid pathways have been explored as potential chemopreventive or therapeutic agents for lung cancer. Combinations of COX and 5-lipoxygenase inhibitors have been shown to be more effective in preclinical studies than individual agents [187]. However, the addition of the selective COX-2 inhibitor celecoxib to standard chemotherapy did not improve survival of lung cancer patients [188]. In a separate study, neither a COX-2 nor a 5-lipoxygenase inhibitor given with standard platinum-based chemotherapy increased overall survival compared to chemotherapy alone [189]. However, in patients expressing high levels of COX-2, chemotherapy plus a COX-2 inhibitor was superior to chemotherapy alone. These data suggest that, as shown in other clinical trials for lung cancer using inhibitors of receptor tyrosine kinases, patient selection will be critical in identifying a subset of patients who are likely to respond to inhibitors of eicosanoid pathways. In the case of COX-2, patient selection based on tumor expression of COX-2 would be hypothesized to identify responders. These patients might be considered “addicted to COX-2,” similar to the concept of oncogene addiction [190]. As an alternative approach, noninvasive attempts to measure COX-2 products in lung cancer patients have been developed [191]. Techniques have been developed to measure the major urinary metabolite of PGE2, 11alpha-hydroxy-9,15-dioxo-2,3,4,5-tetranor-prostane-1,20-dioic acid (PGE-M) as a potential biomarker in lung cancer patients [191]. Examination of other eicosanoids in samples from human lung cancer patients has been undertaken. LTB-4 is induced in urine [192], and breath condensate [193] of lung cancer patients increases with progression and correlates with neutrophils in the sputum, suggesting possible utility as a biomarker. Additional trials are examining the role of eicosanoids in adenocarcinomas with mutations in EGFR as oncogenic drivers. In these studies, patient selection involved the use of PGE-M to define subsets of patients. Phase I studies that use the EGFR inhibitor erlotinib in combination with a COX-2 inhibitor have shown promising results [194].
As mentioned above, prostacyclin analogs have shown efficacy in reversing dysplasia in ex-smokers [154]. These data argue for a larger Phase III trial with incidence of cancer as a primary endpoint. As for therapeutic approaches, it is likely that patient selection will be critical in designing this trial. Preclinical studies have indicated that the effects of prostacyclin analogs in chemoprevention are mediated through activation of the PPARγ, and this involves Frizzled 9 (Fzd9), a canonical receptor for Wnt ligands [195]. Expression of Fzd9 engages an anti-tumorigenic signaling pathway in human NSCLC either through the ligand Wnt7a [196, 197] or through prostacyclin analogs such as iloprost . Thus, elevated expression of Fzd9 in dysplastic lesions may represent a biomarker for chemoprevention by prostacyclin analogs .
Perspective and Future Directions
The relationship between eicosanoid production, inflammation, and cancer has been established for many years. A large number of studies have implicated individual eicosanoids in preclinical models of cancer, using both in vitro approaches and mouse models . These data demonstrate effects both on tumor cells and cells of the microenvironment (see Table 6.1). Nevertheless, we still have a poor understanding of how this complex pathway is integrated into lung cancer development, both in the setting of tumor initiation, as well as in progression and metastasis. This is due to the fact that the eicosanoid pathway is extremely complex. Over 100 distinct products have been identified, and the role of many of these products has not been well studied in the setting of lung cancer, or in fact in any cancer. Many of these products can act on multiple receptors, with potentially opposing effects. Secondly, specific eicosanoids are produced by different populations of cells in a spatiotemporal fashion. Thus, a particular product, such as PGE2, can be produced by both tumor cells and inflammatory cells such as macrophages (see Fig. 6.2). Levels of production will be linked to expression of enzymes in the pathway, the local environment of the cell, as well as factors and interactions with surrounding cells, which control PGE2 production. Since eicosanoids such as PGE2 act locally, it is not known whether production by macrophages vis a vis cancer cells results in similar biological responses. The use of pharmacological inhibitors, which will target all cells, will not be able to address this issue. Thus, we lack a clear picture of how eicosanoid production by specific cell types, as a function of time during tumor initiation and development, contributes to disease progression.
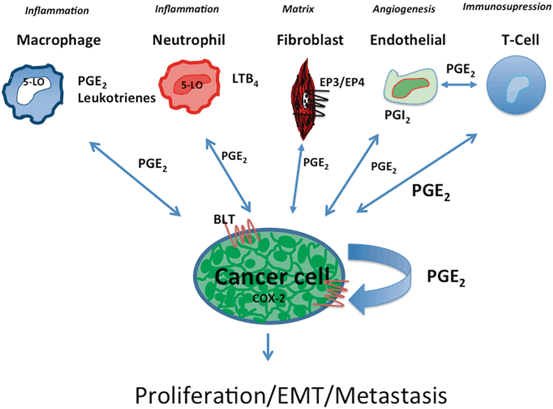
Fig. 6.2
Eicosanoids in crosstalk between cancer cells and the microenvironment. Eicosanoids play a critical role in crosstalk between cancer cells and cells of the TME. At least a subset of lung cancer cells (enriched in adenocarcinoma) produce high levels of PGE2. This can act in an autocrine manner by binding to cell surface receptors on the cancer cells which express G-protein coupled (7-membrane spanning) PGE2 receptors. Alternatively, PGE2 can act on various cells of the microenvironment including neutrophils, macrophages, fibroblasts, endothelial cells, and immune cells. This production can alter the phenotype of each of these cells. In addition, many cells of the environment can produce eicosanoids themselves, which in turn act back on the cancer cell, or on other cells of the TME
Table 6.1
Representative roles for eicosanoids in lung cancer
Eicosanoid | Producing cell | Biological effects | References |
|---|---|---|---|
PGE2 | Tumor cell | Proliferation | |
Macrophage | Cytokine production | ||
Dendritic cells | Antigen presentation | ||
T cells | Immunosuprression | [135] | |
PGD2z | Eosinophils | ||
Tumor cell | Angiogenesis | [37] | |
PGI2 | Epithelial cell | Inhibits tumor initiation | |
TxA2 | Tumor cells | Proliferation | [163] |
Inhibition of apoptosis | [164] | ||
Macrophages | Recruitment | [166] | |
LTB4 | Neutrophils | Recruitment | [176] |
5-HETE | Tumor cells | Proliferation | |
15-(S)-HETE | Tumor cells | PPARγ activation | [75] |
There are precedents for cell specific production of signaling molecules mediating disparate effects. In breast cancer, it has been shown that both tumor cells and macrophages produce the angiogenic factor VEGF. Elegant studies by Johnson et al., using targeted knockout mice, have shown that ablation of VEGF by macrophages resulted in a less tortuous tumor vasculature, which actually promoted tumor growth, whereas deletion of VEGF in tumor cells decreased tumor growth [198]. Interestingly, ablation of VEGF production in macrophages did not alter the overall levels of VEGF in the tumor. These data argue that production of a soluble factor by a specific cell type has a distinct biological response, independent of effects of overall levels of the factor. We would propose that a similar situation will exist for individual eicosanoids. Thus, it will be critical to develop approaches that allow the spatiotemporal determination of individual eicosanoids within developing tumors. Such an approach has been used with MALDI to obtain information regarding the distribution of membrane phospholipids in heterogeneous tissues such as brain [199]. The abundance of eicosanoids is currently too low to apply this technology, but new approaches indicate that this may be feasible in the near future [200]. These tools would provide a deeper understanding of the dynamic nature of eicosanoid production. In preclinical studies, it will also be important to validate the functional role of individual eicosanoid products. This will require the use of global and target knockout mice for both enzymes in the pathway, as well as putative receptors. Fortunately, many of these mice are available, but have not been examined in the setting of lung cancer. For example, in the 5-lipoxygenase pathway, 5-lipoxygenase knockout mice and FLAP knockout mice are available [173, 201]. In addition, mice with deletions in downstream enzymes in leukotriene synthesis have been developed. These include LTA4 hydrolase knockouts [202], which will have an impairment in the production of LTB4, and LTC4-synthase knockout mice [203], which will be defective in production of LTC4, LTD4, and LTE4. Studies employing these animals, along with potential bone marrow transplants will be critical in defining specific 5-lipoxygenase products, as well as the cell of origin .
An additional critical issue is the relationship of the cancer cell to the nature of the inflammatory response. As discussed earlier, a large number of studies over the past 5 years have subdivided cancers according to distinct mutations in critical oncogenes. In lung cancer, adenocarcinoma and squamous cell carcinoma can be classified according to the specific driver mutations, e.g., K-Ras, EGFR, etc. Defining these drivers has allowed the development of targeted therapies and criteria for patient selection in clinical trials and chemotherapy. However, the focus on inflammation and the role of eicosanoids specifically in relation to inflammatory and immune cells has not integrated this approach. Thus, it is not known whether there are distinct roles for specific eicosanoids produced by cells of the microenvironment that depend on the nature of the mutations in the tumor cells. Thus, it is not known whether PGE2 or other eicosanoids produced by inflammatory cells play the same role in K-Ras lung tumor vis a vis and EGFR-mutant lung tumor. Approaches using preclinical models are needed to correlate changes in eicosanoids with tumors driven by different oncogenic drivers. This approach may reveal changes in eicosanoid production that may actually be biomarkers for a particular tumor subtype.
In conclusion, eicosanoid biology has existed for over 50 years, and there has been a large body of work implicating these molecules in cancer. In surveying the literature, it appears that there are waves of interest in this topic, followed by periods of less activity. It is thus somewhat surprising; that there is still much to learn regarding how these molecules contribute to cancer biology. This is particularly true for products of the lipoxygenase and cytochrome P450 pathway. In addition, less is known regarding the contribution of these molecules to molecular crosstalk between cancer cells and TME. From a translational view, these pathways represent easily “targeted” therapies. Thus, a better understanding of the role of individual products is likely to lead to new therapeutic strategies, both in lung cancer and other diseases.
Related posts:
 Inflammation and Lung Cancer: Molecular Pathology
Inflammation and Lung Cancer: The Role of Epithelial–Mesenchymal Transition
Inflammation and Lung Cancer: Addressing Inflammation with Immunotherapy
Inflammation and Lung Cancer: Molecular Pathology
Inflammation and Lung Cancer: The Role of Epithelial–Mesenchymal Transition
Inflammation and Lung Cancer: Addressing Inflammation with Immunotherapy
 Inflammation and Lung Cancer: The Link to Angiogenesis
Inflammation and Lung Cancer: Prevention
Inflammation and Lung Cancer: The Link to Angiogenesis
Inflammation and Lung Cancer: Prevention
 Inflammation and Lung Cancer: The Relationship to Chronic Obstructive Pulmonary Disease
Inflammation and Lung Cancer: The Relationship to Chronic Obstructive Pulmonary Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


