Inflammation and Aging: Introduction
In recent years, multiple studies of older adults have identified strong relationships between serum markers of inflammation and frailty, worsening chronic disease, disability, and mortality. Although these studies are not proof that the chronic activation of inflammatory pathways causes these adverse health care outcomes, recent biological evidence supports that chronic exposure to inflammatory mediators leads to alterations in multiple physiological systems. These in turn contribute to the older adult’s vulnerability to adverse health outcomes. Because the activation of inflammatory pathways appear to be so intricately linked to many aging systems, some investigators have propagated use of the term “inflammaging” as a summary of the physiological and molecular changes consistent with the aging process that are known to be associated with chronic activation of inflammatory pathways. Other investigators now utilize “chronic inflammation” as a descriptor for the ongoing activation of the innate immune system that has been observed in some older individuals. The rising use of these terms in aging research and in clinical practice signifies the emerging importance of inflammation as an important contribution, adverse health outcomes in older adults. The study of the clinical utility of relevant inflammatory biomarkers, the understanding of the specific age-related mechanisms that activate and sustain chronic inflammation in older adults, and exploration of the impact that inflammatory mediators and pathway activation have on specific physiological systems and on the overall vulnerability observed in older adults are certain to impact both research and clinical agendas for older adults in the coming years. Given this crucial focus and the importance of understanding this emerging area of investigation for those who care for older adults, and the high likelihood that both investigators and clinicians will likely encounter this important and evolving area of aging research in the coming years, this chapter will (1) describe evolving definitions of inflammation used in the context of aging research and clinical practice, (2) provide a review of the robust relationships between activation of inflammatory pathways and adverse health outcomes previously identified in older adults, (3) describe the pertinent areas of the molecular biology of inflammation necessary to understand how and why older adults are more vulnerable to inflammatory pathway activation, (4) characterize the relationships between inflammatory pathway activation, multisystem decline, and the biological vulnerability observed in older adults, and (5) highlight the current and potential future clinical relevance of inflammation in older adults.
Inflammation in the Context of Aging and Adverse Health Outcomes
Inflammation, often referred to as the activation of the innate immune system, is a complex and important physiological response to external threats. In general, it is a critical housekeeping function that acts to fight acute infections and repair wounds through common biological pathways that in turn activate hormonal, thrombotic, and cytokine pathways (Figure 4-1). These pathways function throughout the life span to attenuate or eliminate countless infections and injuries from becoming life-threatening events. This inflammatory signaling is in general a self-limiting process that ends when the infection or injury is resolved and the local and systemic inflammatory pathways return to a state of inactivation. Common clinical examples that lead to the activation of inflammatory pathways and that are attenuated by inflammatory processes at any age include responses to localized superficial injuries such as skin lacerations, localized infections such as urinary tract infections or small abscesses, systemic bacterial infections, and major trauma such as bone fractures or organ injury. In the most superficial processes, inflammation is localized and does not usually result in a measurable systemic response. However, in major infections or injuries, systemic activation of inflammatory pathways result in measurable elevations in circulating inflammatory cytokines and other acute-phase reactant proteins, cortisol, and sympathetic nervous system activation.
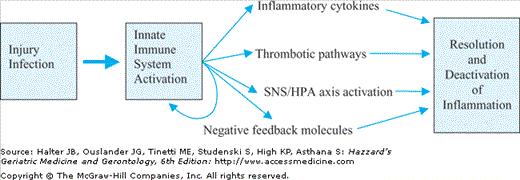
In most cases, these small or large innate immune system responses are largely self-limited by negative feedback mechanisms and by the resolution of initial injury or infection (Figure 4-1). However, recent evidence from population studies of older adults suggests that many older individuals who have no obvious injury or infection have ongoing activation of inflammatory processes even when multiple acute and chronic conditions are accounted for. The inflammatory measurements of total white blood cells, neutrophils, and inflammatory cytokines, even after adjustment for disease states, are usually highly correlated with each other. These studies also provide important evidence of a link between inflammatory pathway activation and adverse health outcomes as detailed below.
Part of the evidence that the chronic activation of inflammatory pathways in older adults may be pathological comes from dozens of well-performed association studies that have identified significant inverse relationships between the common adverse health outcomes of disability, frailty, and mortality and elevated serum interleukin-6 (IL-6) and/or C-reactive protein (CRP). A study by Ferrucci et al. demonstrated a 76% increased risk of developing mobility disability over 4 years in previously nondisabled participants of the Established Populations for Epidemiologic Studies of the Elderly who were in the highest tertile of IL-6 compared to those in the lowest tertile. This study identified a serum IL-6 level of 2.5 pg/mL as an important threshold point beyond which the risk for developing disability rises exponentially. In the Cardiovascular Health Study, investigators identified an odds ratio for being frail of 2.80 in those older adults with a CRP level above 5.77 μg/mL even after excluding those participants with cardiovascular disease and diabetes. In a later study, Puts et al. identified an odds ratio of 1.69 for incident frailty in older adults with a CRP level between 3 and 10 mg/mL. Other investigators have identified strong inverse relationships between inflammatory mediators and mortality in older adults. In the study of osteoporotic fractures, Tice et al. identified an eightfold risk for cardiovascular mortality over 6 years in those with CRP levels above 3.0 mg/mL. Harris et al. identified an 1.9-fold increased relative risk for death over 4.6 years in those participants in the Iowa 65+ Rural Health Study with IL-6 levels greater than 3.19 pg/mL compared to those with low IL-6 levels. Similarly, CRP levels over 2.78 mg/mL also predicted a relative risk for death of 1.6 over the same period of time. Importantly, combining IL-6 and CRP in the analysis increased the sensitivity of identifying those at increased risk of all-cause mortality by identifying a 2.6-fold risk of dying over the same period if both inflammatory markers were elevated compared to the group with neither elevated. Interestingly, many of these studies either excluded participants with severe or chronic disease states known to activate inflammatory pathways, or at least adjusted for them in statistical models, suggesting that this elevation is not purely a result of chronic disease. Although it is not possible to conclude that the activation of inflammatory pathways is causal in these adverse health outcomes for these mostly cross-sectional association studies, their consistency suggests an important connection between chronic inflammatory pathway activation and adverse health outcomes. Further support for the importance of the connection between inflammation and adverse clinical outcomes has come from the many clinical and basic biological studies that have helped to better elucidate the mechanisms by which inflammatory pathways and adverse health outcomes are connected as described below.
Molecular Biology of Inflammation
Clues to etiology logically extend from recently developed knowledge about age-related physiological and molecular changes and from improved understanding of the signal transduction pathways that activate or deactivate the inflammatory process. Although triggers for inflammation may vary, the key gateway molecular signal transduction system centers around the nuclear transcription factor nuclear factor kappa B (NFκB) (Figure 4-2). NFκB, when activated by specific stress or inflammatory signals, facilitates the expression of multiple inflammatory mediators and in general leads to a cascade of molecular messages that in sum represents an inflammation (Figure 4-2, row B). Embedded in this response are connections to other important signals that provide negative feedback necessary to attenuate inflammatory responses, and to other signals that provide connections toward or away from apoptosis. Because of the complexity of this signal transduction pathway, a detailed discussion of this biology is well beyond the scope of this chapter. However, because this biology will likely be relevant to the future clinical care of older adults, it is important to understand how and why these pathways may be chronically active in some older adults, and how their downstream effects contribute to many of the pathophysiological changes and even symptoms that older adults experience. Understanding these molecular pathways also helps to facilitate the understanding of the biology that makes older adults more vulnerable to a host of diseases and adverse outcomes, and may provide some future specificity as investigators begin to target these pathways in pharmaceutical interventions for older adults.
Figure 4-2.

Schematic of the inflammatory gateway nuclear transcription factor NFκB (center circle), along with inflammatory triggers (row A), and inflammatory outflow (rows B and C). Row A shows specific inflammatory stimuli that lead to NFκB activation via specific cell receptors and specific signal transduction pathways (not shown). Activated subunits of NFκB (center circle) in turn leads to the expression of proteins that provide negative feedback for inflammation (not shown) or that propagate inflammatory message (row B) and influence the activation of other pathways (row C) that influence inflammatory response, thrombosis, and the expression of other proteins.
A set of cell surface receptors (Figure 4-2A) act through cell signal transduction pathways to activate NFκB. This links the message of “stress” to the cell nucleus, where inflammatory mediators are in turn generated (Figure 4-2, row B). One of the more important triggers of NFκB-induced inflammatory pathway activators include the tumor necrosis factor alpha (TNF-α). TNF-α is secreted very early in the inflammatory process, usually from immune system cells in response to infections. TNF-α, originally discovered as a serum factor related to cancer, is also secreted in high amounts from the connective tissue surrounding malignancies, and from adipocytes. It has several subtypes of specific cell surface receptors that act to transmit the message of inflammation, which in turn activates NFκB and leads to the expression of a host of inflammatory mediators as described below. Interleukin-1 (IL-1) is most often secreted in response to infections and injury, and activates and amplifies inflammatory signaling through its own cell surface receptor. In addition, IL-1 has recently been identified as a major cytokine that is secreted from senescent cells, providing support that may play an important role in late life activation of inflammatory pathways.7 In addition to these early inflammatory cytokine signals, the bacterial surface antigen lipopolysaccharide (LPS) and some viral particles bind to a family of cell surface toll-like receptors (TLR), which in turn set off a separate signal transduction pathway that in turn amplifies NFκB-related inflammatory signaling. This pathway to inflammatory activation is certainly crucial for the protection of the organism against acute infections, and likely also helps in organized defense against chronic infections. Recent evidence suggests that thrombin, a clotting component, may also activate inflammatory pathways through its own receptor. This provides a link between injury and inflammation. Finally, and potentially very important for aging-related activation of inflammatory systems, the free radicals superoxide and hydrogen peroxide activate inflammatory pathways via signal transduction pathways that facilitate NFκB-mediated inflammatory gene production (Figure 4-2, row A). In sum, all of these specific activating molecules, cell surface receptors, and signal transduction pathways, no matter what the ultimate triggering mechanisms is, transmit the message of stress to the nucleus of the cell, where NFκB-related activity induces the expression of inflammatory mediators that act to either amplify the inflammatory message or exert negative feedback that brakes inflammatory signaling as described below (Figure 4-2).
The molecules that are generated by inflammatory signaling give specificity to the inflammatory message that in sum activates inflammatory pathways or leads to an attenuation of that signal (Figure 4-2
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


