The need to develop sensitive and specific assays amenable to mass screening and automation has become apparent. However test manufacturers are increasingly reluctant to invest resources for screening if the ‘market’ is small, for example screening tests for babesia which may have most impact in a limited region of the Northeaster US, or for outbreaks of chikungunya virus, which are temporally and geographically sporadic. Furthermore, transfusion services are faced with the conflict between increasing test numbers and sensitivity to protect recipients from the risk of acquiring infections by transfusion and, at the same time, increasing the specificity of assays to reduce the number of false-positive results. This is a difficult balance to achieve. More than 10–15% of blood donations are discarded in many countries in addition to the rejection of a similar number of donors before they are bled (Sarkodie et al. 2001). These losses have a major impact on blood transfusion services, many of which are struggling to meet demand. As an added complication, blood components must be held in quarantine while initially positive screening tests are repeated; even in fully computerized transfusion services, the possibility of mistakes with the consequent release of incompletely tested units increases with the number of units held.
Whereas mass screening for T. pallidum with a surrogate assay was introduced in the US in 1938, the first specific screening tests were developed to detect viral antigen (HBsAg) and antibody (HIV, HCV). The confirmation of antigen assays by neutralization of reactivity using specific antibody is more reliable than the open ‘confirmatory’ techniques available for antibody assays. In an effort to reduce the risk of transmitting infection to the lowest possible level, molecular techniques to detect the microbial genomes are used widely for HBV, HCV, HIV and WNV. For ease and cost, pools of 16–90 specimens are screened, but as combination tests evolve, testing of individual donations for each agent has become standard practice. The principles for most of the commonly used screening assays are described below.
Passive Haemagglutination or Particle Agglutination
Standardized antigen or antibody is used to coat tanned red cells or particles such as gelatin or latex. If the specific, complementary antibody or antigen is present in the serum under investigation, the red cells or particles are agglutinated. This type of assay is usually performed in U- or V-well microplates that can be centrifuged to enhance agglutination. The assay is also amenable to automation in blood grouping machines. If antibodies are used to coat the cells for agglutination by antigen in the serum under investigation, the term ‘reverse passive agglutination’ is used.
Enzyme-Linked Immunosorbent Assay
Standardized antigen (e.g. HIV antigen) is linked to a solid phase in the form of beads, the wells of microplates or dipsticks, etc. If antibody (e.g. anti-HIV) is present in the test sample (donor’s serum), which is generally diluted according to the manufacturer’s instructions, it will adhere to the solid phase. The coated wells or beads are then washed; if antibodies are present, they will be detected with an enzyme-linked human antiglobulin reagent, which will, in turn, be detected by the appropriate chromogenic substrate (Figure 16.1a). The antigens can be: (1) disrupted microbial agents with different grades of purity and of contamination with cells in which they are cultured; (2) recombinant polypeptides obtained by molecular genetic methods; or (3) synthetic antigens. The enzymes most widely used are horseradish peroxidase and alkaline phosphatase. The sensitivity of the enzyme-linked immunosorbent assay (ELISA) can be enhanced with biotin–avidin or biotin–streptavidin, or by amplification through the addition of NADP, alcohol dehydrogenase and diaphorase.
Radioimmunoassay
The basic principles are the same as for ELISA, but the label for the antiglobulin is a radioisotope, such as 125I.
Chemiluminescence Assays
The assay is similar to an ELISA but the horseradish peroxidase reacts with luminol in the presence of an enhancer to produce light emission.
Competitive Enzyme-Linked Immunosorbent Assay or Radioimmunoassay
Known antigen (for example HIV-1) is linked to the solid phase and any antibody in the serum will compete for binding with enzyme-labelled or radiolabelled antibody (Figure 16.1b). The more antibody in the sample, the weaker the enzyme reaction available to colour the substrate, or the weaker the radioactive signal. In a competitive assay, the weaker the reaction, the larger is the amount of antibody present, whereas in the antiglobulin assay, the stronger the reaction, the larger is the amount of antibody present.
Sandwich Enzyme-Linked Immunosorbent Assay or Radioimmunoassay
Antigen molecules coat the solid phase as in the antiglobulin ELISA and any antibody present in the test sample will be detected by the reaction with labelled antigen (Figure 16.1c). For the detection of viral antigen, two types of monoclonal antibodies can be used in ‘one-step’ assays, reacting with different epitopes of the antigen to avoid blocking the sites which attach the antibody onto the solid phase (Figure 16.1d).
Sandwich Particle Assay
Particles in suspension are coated with several molecules of antigen. When antibody is present in the test sample, the particles agglutinate (Figure 16.1e).
Screening Assays: Testing Strategy
If donor serum or plasma reacts with a screening assay, most tests are repeated in duplicate. If at least one of the repeat tests reacts, the specimen is considered ‘reactive’. The original sample should then be tested with a ‘supplementary’ or with a ‘confirmatory’ assay using a methodology different from that used in the screening assay. The most definitive type of confirmation is that used for viral antigen and consists of neutralization or inhibition of the antigen–antibody reaction by a well-authenticated antibody.
Western Blot
Lysed viral or recombinant antigens, for example of HIV, are subjected to electrophoresis in sodium dodecyl sulphate (SDS) polyacrylamide gel. The viral polypeptides are separated by migration on the gel, according to their molecular weight. The polypeptide pattern on the gel is transferred (‘blotted’) on to nitrocellulose paper, which is dried and cut into strips. Dilutions of the serum samples found to be positive in screening assays are incubated with the strips; if antibodies to HIV antigens are present, they will combine with different and precise sections of the strip carrying the specific polypeptides. As in any other antiglobulin assay, the strips are washed and antigen–antibody reactions are detected by an appropriately labelled antiglobulin reagent. Antiglobulin is usually labelled with enzyme and a positive reaction is detected by the reaction of the enzyme with its substrate. Antibodies to defined polypeptides can be detected according to the position of the bands on the strip. Criteria of a positive reaction should be well defined for each agent for which Western blot is used as the confirmatory test. Unfortunately, contaminating proteins from viral lysates may travel to positions that are the same as those of specific viral antigens and may sometimes bind to crossreacting antibodies, leading to false-positive reactions.
Recombinant Immunoblot Assay (RIBA)
This assay is used for confirmation of the presence of HCV antibodies. Recombinant and synthetic antigens on a nitrocellulose strip are incubated with the test serum; specific antibodies react when present in the serum and the reactions are visualized as bands.
Nucleic Acid Testing Technology
The principle underlying nucleic acid testing (NAT) is the polymerase chain reaction (PCR), a technique that makes it possible to amplify a short specific sequence of viral deoxyribonucleic acid (DNA) in a sample (see Colour Plate 16.1) (Mullis and Faloona 1987). Ribonucleic acid (RNA) viruses can also be used after treatment with reverse transcriptase. For PCR it is essential to know the sequence of bases flanking the target DNA region. Two specific short fragments of synthetic DNA (oligonucleotide primers) are then synthesized to match (hybridize) the sequences at either end of the target from the 5′-end (see Colour Plate 16.1). The first primer is a copy of the end of the coding strand and the second primer is a copy of the end of the non-coding homologous strand. DNA polymerase is essential in PCR for the synthesis of the complementary strand of a DNA sequence in the presence of the appropriate primers and deoxyribonucleotide triphosphates (dNTPs). A thermostable polymerase enzyme from the bacillus Thermus aquaticus (Taq) has simplified the procedure considerably, as it is relatively unaffected at the denaturation temperature of 94°C and does not need to be replenished at each cycle. The PCR mixture contains the specimen under study, excess oligonucleotide primers, Taq polymerase and abundant dNTPs as well as electrolytes such as Mg2+. The binding of the primers to the flanking sequences of the target DNA (annealing) is possible after separation of the two strands of DNA by melting at 94°C and cooling at 40–55°C. At its optimal temperature of 70–75°C, the Taq polymerase will stimulate the synthesis of complementary DNAs (extension) to the coding and non-coding strands starting from the annealed primers in the 5′ to 3′ direction. The whole process of melting, cooling, reassociation or annealing and synthesis takes 2–3 min and is repeated cyclically. As more than 30 cycles are undertaken per procedure, the target DNA becomes amplified exponentially because the new strands, as well as the old ones, become templates for the excess primers (Colour Plate 16.1). The end result is that the target DNA sequence is amplified 105–106 times in just a few hours. The amplified sequences are detected by electrophoretic separation or by hybrididization to radiolabelled or enzyme-tagged probes. PCR cycling has been automated using microprocessor-controlled heating blocks. Commercial systems can detect fewer than 100 viral copies in plasma or cell samples (Hawkins et al. 1997; Nolte et al. 2001). The major problems with PCR relate to the specificity of the oligonucleotide primers and to the possibility of amplification of non-target sequences that can occur, especially in contaminated samples. Cross-contamination with foreign DNA during PCR assays is by far the greatest problem because the contaminating DNA can also be amplified.
NAT involves more than one kind of assay, but the general principles are similar. The NASBA technique amplifies RNA, based on the principles of bacteriophage replication, with no requirement for thermal cycling; the amplified RNA is detected either by an enzyme-linked assay or by luminometry (Romano et al. 1996). Other amplification techniques include transcription-mediated amplification (TMA) (Sarrazin et al. 2000), the ligase chain reaction for amplification of DNA and the branched DNA signal amplification assay (Allain 2000). Equipment providing varying degrees of automation has been applied to these techniques, and full automation is close to licensure.
Current practice involves two different approaches to NAT screening: plasma pool and single-donation testing. Pool testing costs less, but suffers some loss of sensitivity. Single-unit testing is not yet fully automated and will be more costly unless multiplex testing can be used (Allain 2000).
Hepatitis B Virus
Despite the dramatic reduction in risk of viral transmission during the past three decades, viral hepatitis remains a serious complication of transfusion worldwide. The discovery in 1968 that the viraemic phase of serum hepatitis (hepatitis B) could be recognized serologically (Blumberg et al. 1968; Prince 1968) sparked the hope that all infectious donors could be identified and that post-transfusion hepatitis could be eliminated. However, most transfusion-transmitted cases of hepatitis proved to be associated with viruses other than B (or A), and several other viruses, most importantly HCV, were eventually discovered.
HBV, a double-stranded DNA virus, remains a major human pathogen that causes acute and chronic hepatitis, cirrhosis and hepatocellular carcinoma (Ganem and Prince 2004). An estimated 350 million carriers are found worldwide, and about 1 million deaths a year are attributed to this disease. HBV is an enveloped virus that replicates via reverse transcription of RNA intermediates to translate proteins important for diagnostic tests, HBsAg, HBc, and HBe. The virus is readily transmissible perinatally, by sexual contact, and by needlestick. However, the risk of HBV transmission to transfusion recipients is extremely low when sensitive tests capable of detecting less than 1 ng/ml for HBsAg, and tests for anti-HBc (see below) are used routinely (Biswas et al. 2003). Nevertheless, cases in which HBV is transmitted by HBsAg and anti-HBc-negative donations still occur (Allain 2004). So-called ‘occult hepatitis B virus’ (OBV) may reflect viral mutations responding to humoral and cellular immune pressure (Candotti et al. 2008). The transfusion risk of HBV in the US has been estimated to be 1 in 205 000 per unit (Dodd et al. 2002).
The outcome of HBV infection is heavily influenced by patient age. In the Far East, HBV is acquired more commonly by perinatal transmission than it is in the West. The neonatal immune system appears to tolerate the virus which leads to chronic carriage, lifelong in more than 90% of patients, but rarely in acute hepatitis. A low level of hepatic injury occurs over time. By contrast, immunocompetent adults infected by transfusion or sexual transmission mount a potent cellular immune response that may result in clinically severe hepatitis, but also results in clearance of the infection in 99% of patients (Hoofnagle et al. 2007).
HBV-Associated Antigens and Antibodies: the ‘B-Markers’
Hepatitis B surface antigen (HBsAg, formerly Australia antigen) is unassembled viral coat. Electron microscopy of concentrated serum containing HBsAg reveals the presence of so-called Dane particles, diameter 42 nm, which are known to be the complete virus (Dane et al. 1970). The particles are made up of an inner protein core (HBc) containing partially double-stranded DNA with a single-stranded region of variable length, protein kinases and DNA polymerase, surrounded by an outer coat of HBsAg (see Colour Plate 16.2). HBsAg is shed into the plasma in large quantities in the form of spheres and rods (diameter 18–22 nm). Antibodies to the surface antigen and the core antigen are known, respectively, as anti-HBs and anti-HBc. The major polypeptide in the core of HBV, p19, carries the antigenic determinants of HBcAg and HBeAg (Takahashi et al. 1981). HBseAg is a soluble protein (although also particle associated), found only in some sera containing HBsAg and thought to be a cleavage product of HBcAg. HBeAg in serum reflects viral replication and its presence is associated with infectivity.
In the partially double-stranded DNA, the long (L–) strand is of fixed length, whereas the length of the short strand (S+) is variable. The circular HBV genome is about 3200 nucleotides long. The L– strand carries virtually all the protein capacity of HBV in four open reading frames (ORFs) termed S/pre-S, C, P and X, which overlap one another. The S/pre-S region codes for the envelope proteins and is divided into: (1) the S gene coding for the major protein, HBsAg, 226 amino acids long, carrying the ‘a’ immunodominant determinant, with allelic variations for d/y and w/r (see below); (2) the pre-S2 region, coding for the pre-S2 antigen, a very immunogenic sequence of 55 amino acids, resistant to denaturation, which elicits neutralizing antibodies and, together with the major protein, codes for the middle protein, a glycoprotein 281 amino acids long; and (3) the pre-S1 region coding for a variable sequence of 108–119 amino acids, depending on the subtype, which is essential for recognition of hepatocyte receptors and, together with the S gene and the pre-S2 region, codes for the large envelope protein of HBV. The C region codes for the core protein and the P region for the DNA polymerase.
Subtypes and Genotypes of HBV
All HBV strains have one antigenic determinant (a) in common, which may be of d or y subtypes: the subtypes (ad and ay) have a further determinant that may be either w or r. The subtypes are useful in epidemiological studies, as their distribution varies geographically.
Eight distinct genotypes, A–H, which vary in geographic distribution have been identified. Pathogenicity d/and response to therapy vary according to genotype. Genotype A is most common in the US and Northern Europe, but it is also found in North Africa and India. Genotypes B and C are prevalent in the Asian-Pacific area. Genotype D is found in the Mediterranean area, the Middle East, and India. Genotypes E–H have been isolated in samples from West Africa, the Americas, and France (Kao et al. 2000; Kato et al. 2004).
HBSAG Carriers
The prevalence of HBsAg varies considerably in different parts of the world and exceeds 15% in some populations in Africa, South-East Asia, China and Latin America. In some countries, such as the US, in which the disease is not endemic, donors with a history of hepatitis after the age of 11 years are permanently deferred from donation. Soon after the introduction of testing for HBsAg, the frequency of positive results in volunteer donors giving blood for the first time was about 0.1%, both in the UK (Wallace et al. 1972) and in the US (Cherubin and Prince 1971). In paid donors the frequency of HBsAg-positive donors was about 10 times greater than in volunteer donors (Walsh et al. 1970; Cherubin and Prince 1971). Although the current risk of paid blood donors remains disputed, an analysis of studies published between 1968 and 2001 concludes that for labile blood components, paid donors have higher rates of infectious disease markers than do volunteer donors (van der Poel et al. 2002) two caveats are important. First, the definition of ‘paid’ relates primarily to ‘professional donors’ who receive money or compensation equivalent to money and does not necessarily include family members or donors who receive incentives such as travel and recognition gifts. Second, additional safety measures for handling plasma donations, and the preparation, purification and viral-inactivation steps employed for the production of plasma derivatives, may render the difference in infectious disease marker rates in donors irrelevant for plasma products.
In donors with previously negative tests who give blood on subsequent occasions, the frequency of a positive test for HBsAg is very low: one in 60 000 in one series in north London. Almost all of the positive tests were due to acute infections in young men (Barbara and Briggs 1981). In north London, up to 1983, about 85% of the HBsAg-positive donations detected by screening were from long-term HBsAg carriers; the remainder came from donors with acute HBV infections (Barbara 1983). In a 6-year study of 1.9 million blood donors at five US collection centres between the years 1991 and 1996, no reduction in the frequency (0.2%) of HBsAg in first-time blood donors could be documented (Glynn et al. 2000). Among US red cell apheresis donors, rates of infectious disease markers (/100 000) were 6- to 13-fold higher than among non-apheresis collections for HIV (3.50 vs. 0.53), HCV (21.84 vs. 1.96), and HBsAg (5.83 vs. 0.44) (Zhou et al. 2010). Most of this risk was attributable to first-time male donors. The residual risk estimates among repeat apheresis donors between 2007 and 2008 were comparable to those among repeat whole blood donors. A donor can transmit HBV for as long as 19 years after a positive test for HBsAg has been detected (Zuckerman and Taylor 1969). Carriers clear their HBsAg at a rate of about 1.7% per annum (Sampliner et al. 1979; Barbara 1983). When HBsAg is eventually lost, it is replaced ordinarily by anti-HBs as a marker of immunity.
More than 70% of apparently healthy British blood donors who are found to be HBsAg positive have normal liver function, as judged by serum aspartate transaminase (AST) and aminotransferase (ALT) levels (Barbara and Mijovic et al. 1978). In a Dutch series a similar figure was observed: in the 30% with abnormal liver function tests (LFTs), when the tests remained abnormal over a period of time, moderate to severe liver disease, as judged histologically, was present in eight out of nine cases. All carriers of HBV were found to be either HBeAg positive (21%) or anti-HBe positive (79%). Abnormal LFTs were found significantly more often in HBeAg-positive carriers than in anti-HBe-positive carriers (Reesink et al. 1980). In countries where HBV is endemic, the proportion of HBsAg carriers who are HBeAg positive is higher.
Immunosuppression may cause recrudescence of latent HBV. In patients in whom anti-HBc is the only detectable HBV marker before immunosuppression, HBsAg may appear after immunosuppressive therapy (Nagington et al. 1977). Activation of latent HBV may be mistaken for post-transfusion hepatitis (PTH) in leukaemic patients or in patients with AIDS who receive blood component therapy. Haemophilia patients infected with HBV are more likely to progress to end-stage liver disease when they are co-infected with HCV and HIV (Goedert et al. 2002).
Anti-HBS
Anti-HBs develops in most people who recover from hepatitis B infection. The presence of the antibody in the plasma marks the end of acute infection and prevents re-infection with HBV. Anti-HBs resulting from HBV vaccination appears to be protective, however breakthrough subclinical infections occur with non-A2 HBV subgenotypes, but cause clinically inconsequential outcomes (Stramer et al. 2011a).
Anti-HBC
Anti-HBc is found lifelong in persons who have been infected with HBV and is a marker of past or current viral replication. In acute infection, high levels of IgM anti-HBc appear during the incubation period after the appearance of HBsAg, persist for 3–4 months and are then replaced by IgG anti-HBc. High titres of IgG anti-HBc can be found in carriers who may sometimes also have low levels of IgM anti-HBc (see below). During the recovery phase of acute hepatitis B, anti-HBc may be present in the absence of HBsAg and anti-HBs. Donations taken at this time (‘window’ period) can transmit HBV (Hoofnagle et al. 1978). In a small proportion of carriers, only anti-HBc can be detected in the plasma. Such subjects may transmit HBV by transfusion (Larsen et al. 1990). In countries such as the US, where screening of blood donations for anti-HBc has been introduced, the incidence of PTH due to HBV may have decreased considerably compared with that in countries where routine screening for anti-HBc is not mandatory. Direct testing for HBV with multiplex NAT has eliminated some of this discrepancy. The presence of both anti-HBc and anti-HBs confirms immunity to HBV, whereas their absence suggests susceptibility. However, passive transfer of these antibodies may follow transfusion or childbirth.
HBEAG and Anti-HBE
HBeAg is a marker of infectivity associated with the a high level of HBV replication found during the incubation period and the acute phase of clinical hepatitis B. Anti-HBe develops during recovery. HBsAg carriers initially have HBeAg in their blood, together with a high level of HBsAg. This phase may last for a variable period, sometimes measured in years. Approximately 20% of HBsAg-positive blood donors in the UK have HBeAg (Dow et al. 1980; Harrison et al. 1985), compared with 50% in Mediterranean countries (Lieberman et al. 1983). The HBeAg-positive phase is followed by a second phase in which anti-HBe replaces HBeAg, with HbsAg often falling to lower levels. Although infectivity is greatly reduced during this second phase, blood transfusion can still transmit HBV. Some HBV mutants that are incapable of synthesizing HBeAg have been shown to induce fulminant hepatitis (see section on HBV variants, below).
DNA Polymerase
DNA polymerase is a marker of viraemia. Biochemical assays for the detection of DNA polymerase have been developed, but the assay is not as sensitive as that for detecting HBV-DNA and therefore not useful for mass screening of blood donors.
Serum HBV-DNA
HBV-DNA can be detected in serum by molecular hybridization techniques and PCR; however, PCR is more sensitive by several orders of magnitude than direct hybridization. A low level of HBV-DNA appears before HBsAg at the time of infection and remains detectable in serum and liver tissue in some patients who clear HBsAg from either acute self-limited or chronic HBV infection, or even after a successful anti-HBV treatment (Hu 2002). Occult, silent or latent HBV infection, defined as the presence of HBV infection with undetectable HBsAg, is well documented, although the prevalence of this clinical entity is unknown (Thiers et al. 1988; Allain 2004). HBV-DNA was detected in 8–12% of healthy Chinese people who were positive only for anti-HBc or anti-HBs, and in some healthy Chinese people without any serological HBV markers. On the other hand HBV-DNA was detected in only one-third of HBsAg positives (Pao et al. 1991). The viral load in occult infections is <1000 IU/ml, so that pooled testing would improve blood safety minimally. If blood donors were to be screened for HBV-DNA in addition to HBsAg, the number of extra infectious donors detected would be small in countries with a low prevalence of HBsAg, and increasing immunization together with effective treatment minimize the public health benefit of such screening. A US study with a triplex nucleic acid test of 3.7 million blood donations detected 9 potentially infectious HBV donors (HBV DNA-positive but negative for HBsAg and anti-HBc, 1 in 410 540 donations), including 6 samples from donors who had received the HBV vaccine who likely posed little if any transfusion risk. (Stramer et al. 2011b) Whether screening blood donors in populations with a very high prevalence of HbsAg, such as the Chinese (25%), would be of value, is another matter.
Screening Tests for HBSAG in Blood Donations
The first tests used, such as immunodiffusion and counterimmunoelectrophoresis, were soon replaced by the more sensitive immunoradiometric assay (RIA) and the ELISA, which detect less than 0.5 ng of HBsAg/ml. However, taking the molecular weight of HBsAg as 3 × 106, 0.5 ng is equivalent to 1 × 107 particles. Thus, with a negative result of these tests, as many as 4–10 × 107 antigen molecules may be present per millilitre. Complete prevention of post-transfusion hepatitis B probably cannot be achieved by screening for HBsAg.
Immunoassays are available for the simultaneous detection of HBsAg and anti-HBs. Several monoclonal HBs antibodies directed against the a antigen, common to all HBV subtypes, have been produced and used in sensitive screening assays. However, mutations in the S region of HBV have led to a-deficient ‘escape’ mutants, which could not be detected by assays based solely on monoclonal anti-a (Carman et al. 1990). These mutants are not neutralized by anti-HBs in vivo or in vitro. In addition, if polyclonal assays based on anti-ad are used and the mutating virus was originally ay, infectious donors will be missed. The International Standard Unit (ISU, shortened to IU) has been established and is approximately equal to 1 ng of HBsAg/ml. Because of the ready availability of several commercial assays for HBsAg based on monoclonal antibodies, which can detect as little as 0.25 ng/ml, the minimum sensitivity requirement in the UK is 0.5 IU/ml (0.5 ng/ml) (UKBTS/NIBSC Liaison Group 2001).
Transmission of Hepatitis B Virus
HBV is spread by transfusion of infected blood components and by a variety of other percutaneous exposures (accidental needle-stick, shared syringes and needles by drug addicts, contaminated needles used in acupuncture, dentistry or tattooing, and by the sharing of razors). Pathogen inactivation of plasma during the fractionation process has all but eliminated spread by pooled products such as coagulation factor concentrates. HBV can also be transmitted by sexual contact and by close contact between children leading to cross-infection, presumably by blood (e.g. biting) and other infected body fluids. In endemic areas with a high frequency of HbeAg-positive carriers, transmission from mother to infant in the perinatal period is common and often leads to chronic carriage in the infected infant.
In PTH due to the transmission of HBV by blood or blood components, the mean incubation period was found to be 63 days (range 30–150 days) by Gocke (1972) and 73 days (range 39–107 days) by Prince (1975). Highly sensitive assays now detect the presence of viral DNA 2–6 weeks earlier (Biswas et al. 2003). Less than 5% of those infected develop chronic hepatitis (Seeff et al. 1987).
Washing of red cells reduces the viral load and can render units seronegative. However, washing does not eliminate the risk of transmission of HBV. Red cells from blood that is only lightly contaminated with HBV and is washed, or frozen with glycerol and washed, although apparently HBsAg negative, can still transmit HBV to chimpanzees (Alter et al. 1978a) and humans (Rinker and Galambos 1981).
HBV Transmitted by HBSAG-Negative Blood
Carriers with HBsAg below the detection level can transmit HBV by blood transfusion. Patients who were seronegative for all HBV serological markers, but positive for HBV-DNA by PCR, transmitted hepatitis B to two chimpanzees (Thiers et al. 1988). Moreover, subjects infected with HBV may be HBsAg negative owing to point mutations in the pre-core region, which may result in inability to synthesize HBsAg. Fulminant hepatitis developed in recipients of HBsAg-negative blood from donors infected with this mutated virus. In all of these donors, high levels of anti-HBc were present (Kojima et al. 1991). Extremely sensitive assays for HBV-DNA are defining a disparate group of subjects with occult HBV infection (Allain 2004). The frequency of such occult infections depends on the sensitivity of the assays for HBsAg and HBV as well as the prevalence of HBV infection in the population. Small samples of serum and cells containing HBV-DNA, anti-Hbc and anti-HBs (but not HBsAg) from three subjects who had recovered from HBV infection many years earlier failed to infect three chimpanzees followed for 15 months (Prince et al. 2001). However, the infectivity of larger volumes of blood and the prevalence and clinical importance of occult HBV needs further investigation.
In acute infections there are two periods when HBsAg may be undetectable although the subject can transmit HBV: (1) during the early stages of the incubation period when neither HBsAg nor anti-HBc may be detectable (‘diagnostic window,’ see below) and (2) after clearance of HBsAg but before anti-HBs has become detectable ; in this phase anti-HBc, and often anti-HBe, can be detected (Tedder et al. 1980). Testing all blood donations for anti-HBc as well as anti-HBsAg has decreased the incidence of HBV infection by transfusion in the US. About 50% of cases of hepatitis B that could be transmitted by blood from HBsAg-negative donors can be prevented by screening for anti-HBc (Mosley et al. 1995).
Inadvertent transmission of HBV from an asymptomatic subject in the early stages of the incubation period has been described (Rinker and Galambos 1981). The donor’s washed red cells were given to 32 healthy volunteers (to stimulate antibodies) at a time when the donor was HBsAg negative. Between 36 and 76 days later, the donor turned HBsAg positive and subsequently developed hepatitis. Of 32 subjects who received the donor’s blood, 19 developed HBsAg and 14 of these contracted hepatitis. A further nine subjects developed anti-HBs.
Serological Findings in Cases of HBV Hepatitis
In persons exposed to HBV, HBsAg appears first in the incubation period, followed by anti-HBc. HBV-DNA, DNA polymerase, HBeAg and pre-S2 antigen also appear during the incubation period. In acute clinical hepatitis B, HBsAg reaches a peak at the onset of symptoms and then declines during the illness and convalescence, disappearing from the blood in most subjects after a period that varies from a week to several months. Clinical hepatitis B can be a serious disease with approximately 1–3% of acute cases presenting with fulminant hepatitis. Early experience suggested that 5–10% of infected subjects developed chronic hepatitis, but the number appears to be much lower (Seeff et al. 1987). Subjects who recover have anti-HBs, anti-HBc and anti-pre-S2 in their plasma. The carrier state most commonly develops after asymptomatic infections, especially if infection is acquired in infancy. A study of children from Senegal showed that 80% of infants infected in the first 6 months of life became carriers compared with 15% of children infected between the ages of 2 and 3 years (Coursaget et al. 1987). In the UK, where acute HBV infections occur mainly in young adults, about 5% of those infected become carriers (Barbara 1983). Approximately 2 billion people have been infected worldwide with HBV and more than 350 million are chronic carriers. Approximately 15–40% of infected patients will develop cirrhosis, liver failure or hepatocellular carcinoma (Lok 2002).
Management of HBSAG-Positive Subjects
A survey carried out by Alter (1975) indicated that carriers of HBsAg are not commonly a danger to those with whom they come into ordinary social contact (excluding sexual contact). However, infected health-care workers have transmitted HBV to their patients while carrying out invasive surgical or dental procedures. Twelve such outbreaks involving 91 HBV-infected surgical patients who acquired the infection from 11 surgeons and one perfusion technician were reported in England, Wales and Northern Ireland between 1975 and 1990 (Heptonstall 1991). All medical staff should be vaccinated against hepatitis B. In carriers of HBsAg, viral replication as revealed by the presence of HBeAg and HBV-DNA in serum is a useful index of infectivity. However, there are patients with severe liver disease who have HBV-DNA but no detectable HBeAg. These HBeAg-negative variants are due to point mutations in the pre-core region, which prevent the synthesis of HBeAg (Carman et al. 1989; Harrison and Zuckerman 1992).
Protection Against HBV by Antibody
Subjects whose serum contains anti-HBs are protected from HBV infection (Grady and Lee 1975; Seeff et al. 1977). The administration of immunoglobulin prepared from the small proportion of donors with relatively potent anti-HBs was found to reduce the risk of hepatitis in subjects accidentally exposed to HBV infection compared with a control group treated with standard immunoglobulin (Grady and Lee 1975). Standard immunoglobulin, prepared from large pools of unselected donor plasma, contains a titre of anti-HBs too low to be of value in the prophylaxis of hepatitis B (Seeff et al. 1977).
The major indications for hepatitis B immunoglobulin (HBIg) include acute exposure and prevention of graft infection during and following liver allograft transplantation. Following a single acute exposure to HBV, as when blood known or strongly suspected to contain HBsAg is accidentally inoculated, ingested orally or splashed on to open wounds or mucous membranes of non-immune subjects. In such cases HBIg should be given in a dose of approximately 600 IU (for adults) as soon as possible after exposure; the subject should also be vaccinated. When appropriate, a further dose should be given 1 month later (Deinhardt and Zuckerman 1985). Newborn infants in endemic areas should also be given HBIg (see below). HBV recurrence increases morbidity and mortality in HBsAg+ patients undergoing liver transplantation. Therapy with HBIG with or without lamivudine improves HBV-related morbidity and mortality in HBsAg+ recipients of liver transplants (Loomba et al. 2008).
HBV Vaccine
Two types of HBV vaccine are available and have proven to be safe, immunogenic and efficacious; the first was derived from plasma of carriers as a source of surface antigen and the other is a recombinant DNA product synthesized in yeast (Szmuness et al. 1982; Eddleston 1990). Protective antibodies develop in 80–97% of subjects receiving the full immunization course. Protection conferred by either type of vaccine may not be permanent and, in a proportion of subjects, lasts no longer than 2–5 years or even less. However, most subjects appear to be protected for 15 years or more (McMahon et al. 2005). Reinforcing doses may be needed to prevent HBV infection.
Individuals with levels of anti-HBs below 10 IU/l are susceptible to infection, but it is not clear whether previous immunization helps to prevent the development of the carrier state (Eddleston 1990).
Universal immunization of infants against HBV has been recommended in the USA since 1991. Immunization of patients requiring multiple transfusions of blood or blood products, such as patients with haemophilia or thalassaemia and those in need of renal dialysis, as well as health-care workers and medical or dental students exposed to blood, has long been advised by the Advisory Committee on Immunization Practice of the Centers for Disease Control and Prevention (CDC) (www.cdc.gov/nip/publications/acip-list.htm). In areas where HBV is endemic and access to vaccine is limited, subjects at risk should be immunized; infants born to HBeAg-positive mothers should be treated with HBIg, as well as with vaccine (Ip et al. 1989).
HBV Variants
Occasional cases of hepatitis may be caused by variants of HBV that fail to react in conventional screening tests for HBsAg. Sera found negative for HbsAg on routine screening with polyclonal antibodies, but positive with monoclonal antibody as well as positive for HBV-DNA, were shown to transmit hepatitis to HBV-immune chimpanzees (Wands et al. 1986). Other HBV mutants have arisen by point mutations in the pre-core region of the genome; some have caused fulminant hepatitis (Kosaka et al. 1991; Liang et al. 1991) and others have been found in survivors of fulminant hepatitis or in asymptomatic contacts (Carman et al. 1991). The majority of these pre-core mutants do not synthesize HBeAg, which requires intact pre-core and core regions. Hence, there are patients with severe liver disease who have HBV-DNA but no HBeAg in their serum. In such patients there may be continuous viral replication despite the presence of anti-HBe (Tong et al. 1990).
In subjects immunized with HBV vaccine, the neutralizing antibody is directed against the a determinant of HBsAg. Point mutations have given rise to ‘escape mutants’ of HBV with a epitopes that are not neutralized by anti-HBs. If immunized subjects are infected with these mutants, they will have a variant HBsAg in addition to anti-HBs in their plasma (Carman et al. 1990; Harrison and Zuckerman 1992).
Hepatitis D (Delta Virus, HDV)
Delta virus, a 35-nm defective RNA virus found originally in northern Italians, occurs only in HBsAg-positive subjects because it requires HBV as a ‘helper’ virus (Tiollais 1988). The structure and replication of HDV are well described at the molecular level (Taylor 2003). Fewer than 10% of carriers of HBV are co-infected with HDV and the agent appears to be disappearing (Gaeta et al. 2003). Immunization to HBV should hasten its demise. HDV multiplies in the liver and is transmitted by blood and body fluids. The commonest mode of transmission in Europe and the US is by parenteral inoculation, which explains its association with intravenous drug use (Smedile et al. 1982; Rizzetto 1983; Shattock et al. 1985). Anti-HDV has been found in the plasma of infected subjects in Europe, Australia, Asia and America; in the USA it is found in 3.8% of blood donations positive for HBsAg (CDC 1984), but it is found only very rarely in British blood donors (Tedder et al. 1982). Sexual transmission is particularly common in endemic areas of the Far East.
Superinfection of a carrier of HBsAg with HDV is associated with a chronic course of the delta infection in 70–90% of cases and with an increase in severity of the underlying chronic hepatitis (Smedile et al. 1982; Monjardino and Saldanha 1990). Simultaneous infection with HBV and HDV in a previously healthy subject tends to be followed by clearance of both HDV and HBV, although in some instances fulminant hepatitis or a more severe acute course has been reported (Shattock et al. 1985; Reynes et al. 1989; Fagan and Williams 1990). In HDV infection the delta antigen is present in liver and serum; viral RNA and anti-HDV can also be found in serum. As HDV depends upon the host HBV for its viral coat, chronic HDV infection depends on the persistence of HBV.
Screening for HBsAg in blood donors minimizes but does not abolish the risk of PTH delta in HBsAg-positive recipients (Rosina et al. 1985). The simultaneous presence of HDV and HBeAg in the absence of detectable HBsAg has been reported (Shattock et al. 1985). HDV has been transmitted not only by blood and blood components but also by coagulation factor concentrates (Purcell et al. 1985). Prior to universal use of pathogen-inactivated or recombinant clotting factor in the US, 75% of patients with haemophilia A had serological evidence of exposure to HBV, and 13% to HDV (Kumar et al. 1993). No transmission from properly treated plasma has been observed since 1987.
Hepatitis C Virus (HCV) and Non-A, Non-B Hepatitis
Non-A, Non-B Hepatitis
By 1988, three distinct hepatitis viruses had been well characterized: hepatitis A virus (HAV), HBV and HDV. In 1977, the term ‘NANBH’ was coined for those cases of hepatitis not associated with the above three viruses, or to CMV, Epstein–Barr virus or toxic substances (Dienstag et al. 1977). A year later, NANBH was demonstrated to be transmitted by blood (Alter et al. 1978b; Tabor et al. 1978). Subsequent studies have determined, however, that transfusion accounts for only a small fraction of community acquired cases of NANBH (Alter et al. 1990). Hepatitis C virus (HCV), a flavivirus, has been found to be the cause of most NANB cases (Alter et al. 1989a; Kuo et al. 1989) (see below).
Indirect (‘Surrogate’) Markers to Detect Non-A, Non-B Hepatitis
Because the search for the specific agent or agents that cause NANBH was unsuccessful for so long, blood collectors looked for ‘surrogate’ markers to detect infectious donors. Two such markers, the presence of anti-HBc and elevated ALT levels, were proposed as the result of two independent prospective studies conducted in the USA. Both the multicentre Transfusion Transmitted Virus Study (TTVS) and the intramural investigators at the National Institutes of Health (NIH) Clinical Center showed that the incidence of NANB PTH was almost three times greater in recipients of anti-HBc-positive blood than in recipients of anti-HBc-negative blood (Stevens et al. 1984; Koziol et al. 1986). Although the significance of testing for anti-HBc in the prevention of PTH has been much reduced since the introduction of testing for anti-HCV (see below), its significance for preventing hepatitis B remains. In both of the above studies, elevated ALT levels in blood donors were associated with a significantly increased incidence of NANB PTH in the recipients. However, since testing for anti-HCV has become mandatory, the benefit of testing for ALT levels has declined to such an extent that its value in ensuring blood safety is minimal (Busch et al. 1995).
Identification of Hepatitis C Virus
The early efforts to characterize the agent responsible for NANBH indicated that it had a diameter of 30–60 nm, was chloroform sensitive and could be transmitted to chimpanzees, suggesting a small lipid-coated virus. In general, the concentration of the agent in plasma of infected chimpanzees was not high (Bradley et al. 1985). Collaboration between scientists at the CDC in Atlanta, with access to a source of confirmed infectious NANBH material and a group at Chiron Corporation with experience in molecular genetics led to the cloning and identification of the agent responsible for most cases of NANB PTH (Choo et al. 1989).
Structure of Hepatitis C Virus
HCV is a positive, single-stranded, enveloped RNA virus. The size of its RNA varies in different isolates but is about 10 kilobases long. Its genomic organization is similar to that of Flaviviridae and Pestiviruses (Miller and Purcell 1990). The genome is approximately 10 000 nucleotides and encodes a single polyprotein of 3010–3011 amino acids which is subsequently processed by host cell and viral proteases into three major structural proteins and several non-structural proteins necessary for viral replication (see Colour Plate 16.3). The structural proteins include the core and the envelope proteins E1 (glycoprotein 33/35) and E2/NS1 (gp72). The non-structural proteins comprise the N2–N5 proteins, the function of which is not entirely clear; however, the region does code critical enzymes such as an NS3 helicase and an NS5 polymerase (Takeuchi et al. 1990; Choo et al. 1991; Weiner et al. 1991).
At present, HCV genotypes and more than 100 subtypes have been described (Simmonds 2004). The distribution of the subtypes varies in different geographical areas. The genotypes are of clinical importance and clearly influence response to antiviral therapy (Ghany et al. 2010). HCV type II (subtype 1b), for example, is associated with a low response to therapy with alpha-interferon and with relapse after treatment (Yoshioka et al. 1992; Brouwer et al. 1993).
Evidence That Hepatitis C Virus Is the Main Agent of Non-A, Non-B Post-Transfusion Hepatitis
Using the first available test for anti-HCV, all of 15 well-characterized patients with NANBH had detectable antibody, but only three out of five patients with acute resolving NANBH gave positive results. HCV antibodies were detected in six out of seven NANBH sera infectious for chimpanzees (Alter et al. 1989). Similar results were obtained in Japan and Italy (Kuo et al. 1989). Furthermore, anti-HCV was present in all of 27 haemophiliacs who had been transfused at some stage with untreated clotting factor concentrates, but in none of 28 patients who had received only products subjected to viral inactivation procedures (Skidmore et al. 1990). Sensitive and specific anti-HCV assays, together with NAT tests, have confirmed the primacy of HCV as the NANB agent; however, evidence from multiple episodes in single patients and chimpanzee cross-challenge studies suggests the presence of additional agents (Mosley et al. 1977; Yoshizawa et al. 1981).
The incidence of PTH has significantly decreased since screening for anti-HCV was introduced. Whereas 38 (12.4%) of 226 patients who underwent cardiovascular surgery and received blood transfusions developed PTH before screening was begun, none of 87 patients developed PTH after receiving anti-HCV-negative blood (Wang et al. 1994). In the 40-year prospective studies at the NIH, PTH rates fell from more than 30% in the 1960s to <1% in 1995 and close to zero by 1997 (Alter and Houghton 2000).
Clinical Course of Hepatitis C
According to the WHO, approximately 180 million individuals worldwide are chronically infected with the hepatitis C virus (HCV) and are at risk for related cirrhosis and hepatocellular carcinoma. Most cases of hepatitis C are mild and non-icteric, but 50–70% of the cases become chronic and present as chronic persistent hepatitis or chronic active hepatitis. About 20% of patients infected with HCV develop cirrhosis, although the rate is variable and progression to cirrhosis may take 50 years (Ghany et al. 2003). Patients infected with HCV are particularly susceptible to the hepatotoxic effects of alcohol (Corrao and Arico 1998; Harris et al. 2001). Differences in the clinical course of HCV infection in different parts of the world may be due to different genetic types of the virus or, given the chronicity of the infection, to the time of origin of the HCV epidemic in each country (Tanaka et al. 2002).
Prior to viral inactivation of fractionated plasma, some 80% of severe haemophilia A patients, who received numerous infusions of factor VIII derived from pooled plasma donations, developed chronic hepatitis, almost certainly related to HCV exposure (Aledort et al. 1985; Hay et al. 1985). Long-term studies of patients with PTH, most of whom were middle-aged when infected, show no increase in mortality, but a distinct increase in liver-related mortality (Seeff et al. 2001). Cohorts of women of child-bearing age infected with Rh immunoglobulin exhibit low percentages of inflammation and cirrhosis when followed for 17–20 years (Kenny-Walsh 1999). Although HCV infection clearly progresses over time and is exacerbated by alcohol and co-infection with HIV (Goedert et al. 2002), as many as 20% of adults and 45% of children appear to clear the infection spontaneously (Vogt et al. 1999; Seeff et al. 2001).
Screening and Confirmatory Assays for HCV Antibodies
When populations at low risk were screened with the first ELISAs (‘first-generation’ assays) 60% of the reactive samples proved to be falsely positive (van der Poel et al. 1989). The sensitivity of these assays was poor in the early stage of infection (Esteban et al. 1990). The anti-HCV window ranged from 3–6 months and some patients with PTH never seroconverted with this assay. The limited sensitivity of the ‘first-generation’ assay was attributed to the use of the c100/3 non-structural antigen alone, which represents only 12% of the viral genome (van der Poel et al. 1990) (see Colour Plate 16.3).
Second-generation assays, in which additional viral antigens are used, derived from both the structural and the non-structural part of the HCV genome (the c22, c33c and 5.1.1 antigens; Colour Plate 16.3), proved more sensitive than first-generation assays (Aach et al. 1991). In the third-generation assays, an additional non-structural antigen (NS5) is included. These assays are slightly more sensitive than second-generation assays and the percentage of false positives is lower than in second-generation assays (Uyttendaele et al. 1994; Lee et al. 1995).
To address the problem of false-positive reactions, screening strategies confirm reactive samples with a supplemental assay. Recombinant immunoblot assays (RIBAs) with four recombinant proteins (c22-3, c100-3, c33c and 5.1.1) (Leon et al. 1991; Alter 1992) or NAT assays are used. The RIBA is considered to be positive if a reaction occurs with at least two antigens. A reaction with only one antigen is considered to be an indeterminate result. Evaluation of indeterminate results in RIBA-2 showed that these may be specific (HCV RNA positive) or non-specific (HCV RNA negative). The third-generation RIBA (RIBA-3), in which the recombinant c22-3 protein is replaced by a four-epitope synthetic c22 protein and a second NS5 protein is added, has been found to be more sensitive and specific than previous assays (Pawlotsky 1994; Damen et al. 1995). Nevertheless, indeterminate results are still obtained, in which case HCV RNA detection by PCR is mandatory. Recent studies of a cohort of women infected with hepatitis C anti-Rh immune globulin during pregnancy suggests that many ‘false-positive’ RIBA assays do in fact represent exposure to HCV in the distant past when assays of cellular immunity are performed (Takaki et al. 2000). Similar results have been confirmed with a cohort of otherwise healthy blood donors (Semmo et al. 2005). Long-term prospective studies will determine whether these findings indicate that spontaneous recovery from HCV infection is higher than conventionally taught.
Polymerase Chain Reaction for the Detection of Hepatitis C Virus RNA
The most sensitive method for screening for HCV relies on detection of RNA by some gene amplification technique, NAT. NATs turn positive within 3 weeks of infection. HCV NAT using the minipool technique is now used to screen all blood and plasma donors. Although NAT is extremely sensitive and has reduced the HCV window significantly, HCV RNA levels fluctuate during the window period and during the course of chronic infection. High rates of HCV transmission have been found at all levels of viraemia, and donor blood may be infectious even when RNA is undetectable in the TMA assay (Fang et al. 2003; Operskalski et al. 2003). Current HCV RNA testing will not interdict all infectious units, even with single-donation testing, and serological testing remains essential for screening blood donors.
Transmission of Hepatitis C Virus
HCV is transmitted by blood components and has been transmitted by incompletely inactivated plasma fractionation products including IVIG, anti-D Ig for i.v. use and factor VIII concentrate (Yap et al. 1993; Bjoro et al. 1994; Bergman 1995; Meisel et al. 1995; Power et al. 1995). HCV has never been transmitted by albumin concentrates or by anti-D for i.m. use. The risk of transmission from mother to infant is small, although increased when co-infection with HIV exists. The risk is directly related to the amount of virus in the maternal blood, approaching 36% if the viral titre exceeds 106 and almost non-existent at lower titres. The HCV genotype in mothers and infants was identical in nearly all cases (Ohto et al. 1994; Ferrero et al. 2003). The importance of sexual transmission of HCV is controversial. In most studies the techniques used were insensitive and other (mainly shared) risk factors in the sexual partners were not always taken into account. In a study of 50 heterosexual partners of infected subjects over a mean of 13 years, no partner was found positive (Bresters et al. 1993). Among 895 monogamous heterosexual partners of HCV chronically infected individuals in a long-term prospective study, 776 (86.7%) spouses were followed for 10 years, corresponding to 7760 person-years of observation. Three HCV infections were observed during follow-up corresponding to an incidence rate of 0.37 per 1000 person-years. However, the infecting HCV genotype in one spouse (2a) was different from that of the partner (1b), clearly excluding sexual transmission. The remaining two couples had concordant genotypes, but sequence analysis of the NS5b region of the HCV genome showed that the corresponding partners carried different viral isolates (Vandelli et al. 2004). Sexual transmission of HCV within heterosexual monogamous couples must be extremely low. Different results were obtained in China and Japan where 7 out of 38 and 14 out of 195 spouses of infected subjects were found positive (Anahane et al. 1992; Kao et al. 1992). However, the prevalence of HCV in these countries is very high and the results may have been confounded by infection of both partners by an external source. The prevalence of HCV in homosexual men is much lower than that of anti-HBc or anti-HIV (Esteban et al. 1989).
HCV infection does not confer protective immunity (Farci et al. 1993). An important feature of HCV is its high degree of genetic variability, which is due to the inherent low fidelity of the viral replication machinery. HCV circulates in vivo as a population of genetic variants that have been called ‘quasispecies’ (Farci 2001). Quasispecies develop in response to immune pressure and represent an escape mechanism for the virus to evade neutralization and clearance. The clinical significance of quasispecies development in transmission, infectivity and in the natural history of the disease is unclear, but in perinatally infected children, the evolution of HCV quasispecies correlates with hepatic injury (Farci et al. 2006).
Residual Risk of Post-Transfusion Hepatitis: Beyond the Hepatitis Alphabet
Since the introduction of testing for HBsAg, anti-HBc, anti-HCV and HCV NAT, the risk of PTH has been reduced dramatically. Nevertheless, cases of PTH due to HBV and HCV still occur, generally caused by transfusion of blood from donors in the early stages of HBV or HCV infection. In the US, where screening for anti-HBc is done routinely in addition to screening for HBsAg, anti-HCV and HCV NAT, the residual risk of the transmission of HBV or HCV has been calculated as one in 205 000 donations and one in 1 935 000 donations respectively (Dodd et al. 2002). Cases of non-A, non-B, non-C PTH also occur but accurate estimates of frequency are not available.
Hepatitis E Virus
Hepatitis E virus (HEV) is a non-enveloped RNA virus thought until recently to be responsible primarily for epidemic hepatitis in Asia, Africa, Latin America and the Middle East and sporadic hepatitis in developed countries. There are four serotypes, two of which are found primarily in animals such as swine and deer. The agent has been cloned (Reyes et al. 1990; Worm et al. 2000), which led to the development of molecular tests and serologic assays for detecting anti-HEV IgG and IgM (Dawson et al. 1992). Epidemic HEV is nearly always transmitted by the faecal–oral route. However a study of samples from England and Wales, demonstrated that 9% of the hepatitis E cases serologically diagnosed between 1996 and 2003 were not related to recent travel. Similar findings are being reported in the US and France. Molecular analysis found that the infections in these non-travellers were of genotype 3 as contrasted with the genotype 1 and 2 strains found in hyperendemic regions (Ijaz et al. 2005). Surprisingly, the majority of infections occurred in middle-aged, elderly Caucasian males. Some have speculated that infection may be the result of consuming undercooked pork. Whereas the main routes of transmission in such countries remain unclear, like HAV, transient viraemia develops after infection. At least five cases of endemic HEV have been transfusion-transmitted, and the true risk is not known (Matsubayashi et al. 2004; Colson et al. 2007). In Germany, the prevalence of anti-HEV in patients with coagulation disorders has been found to be low (Klarmann et al. 1995). HEV currently appears to be of negligible importance in blood transfusion. However overall HEV seroprevalence estimates of 13.0% and 13.5% were reported from UK cohorts in 1991 and 2004, and recent evidence suggests that a chronic viraemic phase may exist (Ijaz et al. 2009). Sporadic hepatitis E also has been reported in other parts of Europe as well as in the US and in blood donors in Japan (Schlauder et al. 1998; Mansuy et al. 2004; Sakata et al. 2008). The agent is difficult to inactivate and viral sequences have been detected in coagulation factor concentrates.
GB Viruses (GBV/HGV)
Blind molecular cloning, similar to the strategy used to discover HCV, has resulted in the detection of several other transfusion-transmitted viruses. The first agents associated with acute icteric hepatitis that were passaged serially in marmosets came from the blood of a 34-year-old surgeon (GB) (Deinhardt et al. 1967). Later, these agents were characterized extensively and appeared to be distinct from the other hepatitis viruses (Schlauder et al. 1995). Using amplification techniques, cloning and sequencing, at first two viruses, GBV-A and GBV-B, found only in tamarins, and later a third virus, GBV-C, from a human serum, were identified (Simons et al. 1995; Yoshiba et al. 1995). Of the three ‘GB’ viruses, GBV-C seems to be the only human virus. GBV-C has been recovered from the blood of several patients with hepatitis, from multitransfused patients, haemophiliacs and i.v. drug users. The GB viruses are flavivirus-like agents similar to HCV, with a similar genomic organization. However, there is only 26% homology between the GB viruses and HCV. Homology between GBV-A and GBV-C is fairly strong (48%), but between GBV-A and GBV-B and between GBV-C and GBV-B only about 27% (Zuckerman 1996). In relatively small studies, the prevalence of GBV-C has been found to be quite high: 2% in blood donors in the US and the UK and 12–14% in drug addicts and multiply transfused patients. Viral RNA has been detected in 20% of seropositives and viraemia can persist for months or years (Denis et al. 1996). GBV-C has been detected in three out of six patients with fulminant non-A, -B, -C or -E hepatitis but the real significance of the virus as a cause of hepatitis, PTH and chronic liver disease has still to be established (Yoshiba et al. 1995). Lack of a temporal relationship between peak viraemia and raised ALT levels has cast doubt as to whether GBV-C can cause liver disease (Alter et al. 1997). Indeed, GBV-C may not even be a hepatotropic agent.
A virus, provisionally designated HGV, was isolated from the blood of a patient with chronic hepatitis (Linnen et al. 1995). The virus has been cloned and found to have 95% homology with GBV-C but to be only distantly related to HCV. GBV-C and HGV are almost certainly independent isolates of the same virus that can be transmitted by transfusion but do not result in hepatitis.
TT Virus
Using representational difference analysis to compare pre- and post-hepatitis specimens, investigators in Japan discovered a novel non-enveloped, single-stranded DNA virus that correlated with patient elevations of ALT (Nishizawa et al. 1997). TT virus (TTV), named after the initials of the index case, is readily transmitted by transfusion and appears to replicate in the liver (Rodriguez-Inigo et al. 2000). Although 7.5% of the USA blood donor population carries this virus, an almost identical percentage of transfusion recipients (>20%) are infected whether or not they develop hepatitis (Matsumoto et al. 1999). Although TT virus does not appear to be a cause of PTH, a broad range of isolates have now been identified in this family of Circoviridae and it is possible that one of the variants may turn out to be hepatotoxic.
SEN Virus
Another transfusion-transmitted member of the family of Circoviridae was isolated from, and named after, an Italian patient with the initials ‘SEN’ (Tanaka et al. 2001). The prevalence of SEN virus (SEN-V) in 436 volunteer donors was 1.8%. Two SEN-V variants (SENV-D and SENV-H) were unequivocally transmitted by transfusion and associated statistically with transfusion-transmitted non-A to -E hepatitis (Umemura et al. 2001). Even if this association is confirmed, the clinical importance of this agent still remains to be established (Sagir et al. 2004).
Hepatitis A Virus
Hepatitis A virus (HAV), a 27-nm, non-enveloped picornavirus and the infectious agent of epidemic hepatitis transmitted by the faecal–oral route, causes only transient viraemia and does not induce a carrier state. Although the incubation period following infection lasts for several weeks, viraemia lasts only a day or two during acute illness. HAV rarely causes PTH. In 14 published series, relating to transfusions given to about 9000 subjects, not a single case of PTH due to HAV was observed (Blum and Vyas 1982). Transmission of HAV, leading to the development of PTH, can occur in special circumstances (Seeberg et al. 1981; Hollinger et al. 1983; Sherertz et al. 1984). In London, 21% of donors are immune to HAV, whereas almost all subjects in the developing world have been infected by the age of 10 years (Purcell and Ticehurst 1988).
In cases where hepatitis A has followed the transfusion of blood, red cells or fresh-frozen plasma, the donor has been in the early stages of incubating the disease, and then only when the recipient is susceptible to HAV, and when any other units transfused are devoid of anti-HAV that would otherwise neutralize the virus. A single unit of blood transmitted HAV to 11 infants in a neonatal intensive care unit (Noble et al. 1984). Acute infection with HAV is most readily recognized by the finding of specific IgM antibodies to the virus.
An unusual outbreak of transfusion-transmitted HAV occurred in 41 Italian haemophiliacs. The source of infection seems to have been factor VIII concentrate from a local plasma fractionation plant. HAV, with a non-lipid envelope, would be resistant to the solvent–detergent method used for viral inactivation (Mannucci 1992). Further outbreaks of HAV transmission due to solvent–detergent-treated factor VIII and factor IX concentrates have been reported by commercial fractionators, and plasma pools are now routinely screened for HAV with a NAT assay.
In general, normal Ig for i.m. use contains enough anti-HAV to protect travellers to areas endemic for HAV. Now that a hepatitis A vaccine is available, haemophiliacs and frequent travellers have an alternative to receiving repeated injections of normal Ig (Craig and Schaffner 2004).
Human Immunodeficiency Viruses
Retroviruses and Blood Transfusion
Although retroviruses were among the first known viruses, for almost a century they were considered to be ‘animal viruses’ not transmitted to humans. Until 1980, retroviruses had not been linked to human disease, let alone to transfusion-transmitted infection. However, the sensitive immunological, biochemical and molecular approaches that became available in the 1970s led to the discovery of the first human retroviruses, isolated from lymphocytes of a patient with T-cell leukaemia and appropriately named human T-lymphotropic virus type I (Poiesz et al. 1980). Knowledge of the existence of prototype retroviruses encouraged a search for a human retrovirus as the aetiological agent of AIDS. The causative agent, originally referred to as LAV, or HTLV III and now called human immunodeficiency virus (HIV) was discovered in 1983 (Barre-Sinoussi et al. 1983). Numerous isolates of the different human retroviruses have since been reported.
Retroviruses owe their name to their ability to reverse the normal sequence of events in macromolecular synthesis; they are RNA viruses, which, after entering host cells and losing their envelope, use ‘reverse transcriptase’ (RNA-dependent DNA polymerase) together with cell-derived RNA as a primer to transcribe a double-stranded DNA copy of the single-stranded viral RNA genome. The DNA copy enters the nucleus and is permanently integrated as a DNA provirus into the DNA of the host cells, mainly lymphocytes, but also monocytes, macrophages and other cells. The provirus remains latent and replicates as an integral part of the host genome. The synthesis of new viral proteins and viral messenger RNA is programmed by proviral DNA and mediated by host enzymes. The virus consists of two identical molecules of single-stranded RNA, in a protein core containing several molecules of reverse transcriptase and cell-derived molecules. The core is surrounded by an envelope consisting of virally encoded glycoproteins (gps) and host-derived lipids that are acquired when the newly assembled viruses bud out through the cell membrane. The progeny viruses will then infect other cells within the same host (Greene and Peterlin 2002).
All retroviruses contain three main structural genes: the gag gene codes for different molecular weight proteins that are integral to the nuclear core; the pol gene codes for reverse transcriptase; and the env gene specifies the envelope glycoproteins. The primary products of these three genes are larger parent polypeptides that can be enzymatically split into smaller peptides. Human retroviruses, especially HTLV-I and -II, have regions of homology between them (Wong-Staal and Gallo 1985; Gallo 2003). Retroviruses are composed of 60–70% protein, 30–40% lipid (confined to the envelope), 2–4% carbohydrate and 1% RNA. The proteins are immunogenic. The integrity of the envelope is essential for infectivity. The high lipid content of the retroviral envelope renders these agents very susceptible to disruption by detergents and organic solvents.
Human retroviruses all share a tropism for lymphocytes inducing fusion and giant cell formation in vitro and impairing function in vivo. To enter the cell they bind to cell receptors for the retroviral glycoproteins. The high-affinity receptor for HIV has been identified as CD4 on helper T lymphocytes, macrophages and other cells. Accessory cell surface molecules called coreceptors, shown to be chemokine receptors (either CCR5 or CXCR4), are required for fusion with the cell membrane (Berger et al. 1999; Weiss 2002). The HTLV-I and -II virus receptor is the transporter of glucose GLUT1 (Manel et al. 2003). All human retroviruses code for a small crossreactive major core protein, p24, and have similar modes of transmission, i.e. sexual, congenital and by blood or body fluid (Wong-Staal and Gallo 1985).
HIV-1 and HIV-2
The causative agent of AIDS was originally described as LAV (lymphadenopathy-associated virus) (Barre-Sinoussi et al. 1983; Vilmer et al. 1984). Gallo and his colleagues succeeded in culturing the same virus, which they called HTLV-III, in large quantities in continuously replicating T cells (Popovic et al. 1984). This virus, together with visna virus, belongs to the lentivirus subfamily of retroviruses. HIV has a great propensity for genetic variability, especially within the env gene coding for the major external glycoprotein (Wain-Hobson et al. 1985; Wong-Staal et al. 1985), which carries the epitopes that react with the scarce viral neutralizing antibodies. Extreme genetic diversity, with variants classified as group M (main) with subtypes (clades) A–K, group O and group N, has implications for screening, diagnosis and treatment (Stebbing and Moyle 2003).
A second distinct retrovirus named HIV-2 (LAV-2) causes a somewhat milder disease. Infection with HIV-2 occurs mainly in West Africa, but an increasing number of cases have been recognized in Europe, India, and the US. Although this virus is also lymphotropic, cytotoxic and neurotropic, and shares epitopes of the core and pol proteins with HIV-1, HIV-2 nucleotide sequence identity with HIV-1 isolates is only 40–50%. The main differences between HIV-1 and -2 lie within the envelope nucleotides and proteins. HIV-2 has closer sequence identity with the simian retrovirus SIV (Clavel et al. 1986; Brucker et al. 1987; Brun-Vezinet et al. 1987). The different subtypes of HIV-2 are analogous to the different groups of HIV-1 (Damond et al. 2004).
The env gene of HIV-1 codes for a major precursor polyprotein p85. This protein becomes heavily glycosylated to form the exterior glycoprotein gp160, which is processed into gp120 and gp41 by a viral protease (Wong-Staal and Gallo 1985). Gp41 is a transmembrane protein that anchors gp120 in the membrane and has specific amino acid domains for binding to the CD4 molecule on target cells, allowing the entry of the virus into the cell (Colour Plate 16.3; Weiss 2002). The gp41 protein is also important for syncytial fusion of cells (Brun-Vezinet et al. 1987). There is no crossreactivity between the envelope glycoproteins of HIV-1 and -2. The gag gene of HIV-1 codes for the large precursor protein of 55 kDa, p55, which is enzymatically cleaved into the smaller fragments p24, p17 and p15 (Veronese et al. 1988). The p24 protein encloses the two strands of RNA and the reverse transcriptase. The gag proteins p9 and p7, derived from p15, surround the two strands of RNA and form part of the core; p24 and p17 share epitopes with the less well-defined HIV-2 core proteins, p26 and p16 (Brun-Vezinet et al. 1987). The pol gene products of HIV-1 are derived from the large precursor molecule Pr 180 gag-pol and comprise reverse transcriptase, viral integrase and viral protease, the last named being responsible for cleaving both the large Pr 180 gag-pol and the p55 gag precursor into the core proteins (Schochetman 1992; Weiss 2002).
The cell membrane protein CD4 is the main receptor for HIV (Dalgleish et al. 1984). The HIV envelope glycoprotein gp120 binds specifically to CD4-bearing cells and interacts with gp41 for virus–cell and cell–cell fusion events, with the formation of syncytia between infected and uninfected cells. Two molecules act as secondary receptors for HIV-1: one is a G-protein ‘receptor-associated’ molecule CCR-4 (Feng et al. 1996); the other is the β-chemokine receptor CCR-5 (Deng et al. 1996; Dragic et al. 1996). Most viral isolates use CCR-5. Penetration of HIV into the cell occurs by fusion of the viral and cellular membranes. Once the virus enters the cytoplasm, its partial uncoating activates its reverse transcriptase, which converts RNA into double-stranded DNA. Viral DNA is then integrated as provirus into the host DNA. There are other less efficient routes for entry of HIV into cell. Antibody-coated HIV can adhere to monocytes and macrophages via Fc receptors and possibly via complement receptors (Markovic and Clouse 2004).
Once the virus enters susceptible cells, it either remains latent or establishes ‘factories’ of progeny virus in the infected host, so that all subjects who are shown to be infected with HIV should be assumed to have persistent infection and hence to be infectious to others (Schochetman 1992). The cells most susceptible to HIV infection, carrying significant amounts of CD4 receptors, are T-helper lymphocytes, macrophages, monocytes, megakaryocytes and some bone marrow stem cells. Cells carrying low levels of CD4, such as the epidermal Langerhans cells, dendritic cells and certain cells of the central nervous system, are also susceptible. Some cells such as glial cells, colorectal and fetal brain cells are susceptible to HIV infection despite their lack of CD4 receptors (Takeda et al. 1988; Levy 1989).
Course of HIV Infection (Figure 16.2)
For the first few days after infection, no markers of HIV can be detected in blood, an interval known as the ‘eclipse’ phase. Viraemia follows for a period of several weeks, first with intermittent ‘blips’ of virus interrupting periods of undetectable markers in blood. This stage is followed by a ‘ramp up’ phase at about day 10 when HIV viral copy number rises rapidly (Fiebig et al. 2003). At about day 17, p24 antigen becomes detectable in serum and at about day 22, anti-HIV seroconversion occurs. During this period, more than 40% of patients develop an acute, flu-like or mononucleosis-like syndrome (Kahn and Walker 1998). Two to four months after sexually transmitted infection, and 1–2 months after transfusion-transmitted infection, more than 95% of HIV-infected subjects exhibit a wide range of antibodies to the structural env, gag and pol viral proteins, mostly IgG with some IgA and transient IgM at the start of the immune response (Horsburgh et al. 1989). The period between infection and the detection of HIV, the ‘window’ period, has not been as clearly defined after sexual exposure as it has after blood transfusion because the precise date of infection is usually unknown.
Figure 16.2 A schematic, semi-quantitative display of the progression of HIV markers. From top to bottom: WB, Western blot; Ab, HIV antibody; RNA, HIV RNA; LS-Ab, HIV antibody determined by sensitive/less sensitive enzyme immunoassay testing strategy; p24 Ag, HIV p24 antigen, from time of exposure (day 0) through the first 200 days of infection. As each of the markers appears in the bloodstream, the infection is assigned a new stage from 0 (eclipse period, ecl.), characterized by undetectable viral markers in blood samples; reported to last on average up to 11 days from viral exposure through stage I (definitive HIV RNA viraemia), stage II (p24 antigenaemia), stage III (HIV EIA antibody reactive), stage IV (Western blot indeterminate, ‘I’), stage V (Western blot positive without p31 band, ‘P*’) and stage VI (Western blot positive, ‘P’ with p31 band). Stages I–VI were derived from an analysis of the ‘A’ set of plasma donor seroconversion panels as described here. The standardized optical density (OD) cut-off for the sensitive/less sensitive EIA may be varied with recommended cutoffs from 0.5 to 1.0. Cutoffs of 0.5 and 1.0 would result in average demarcations of recent from early chronic infection at 124 and 186 days respectively.
(Source: Adapted from Fiebig et al. 2003. Reproduced with permission of Lippincott Williams & Wilkins.)
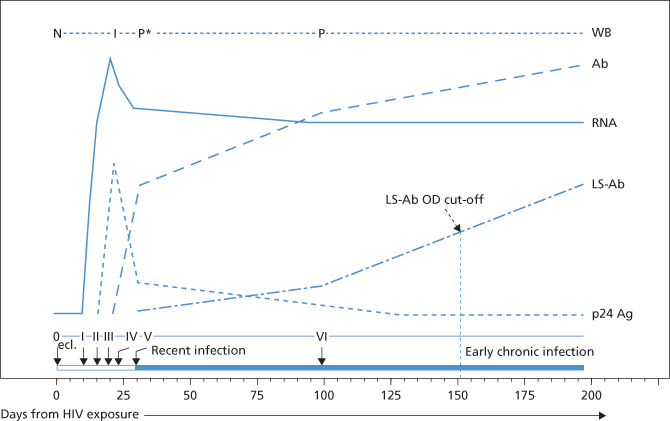
As soon as anti-p24 develops, p24 antigenaemia disappears. A long asymptomatic period follows primary infection; however, disease progression is relentless during this interval (Pantaleo et al. 1993). Antigenaemia may reappear in the late stages of AIDS and intermittently during the asymptomatic phase of infection, which may last for more than 10 years. Levels of all HIV antibody specificities are usually very high during the asymptomatic period but which antibodies combat infection and which if any enhance disease progression is unknown. The cellular response appears to put selective pressure on viral mutation (Soudeyns et al. 1999). Disease develops when the CD4 helper cells have almost completely disappeared, leading to impairment of the immune system and spread of HIV with signs of disease in multiple organs; anti-p24 declines or disappears and p24 antigen reappears at this stage. Other antibodies also decline, with the exception of anti-gp41. CD4 T-cell depletion leads to progressive immunological unresponsiveness to foreign stimuli with increased susceptibility to opportunistic infections (Pneumocystis carinii, CMV, atypical mycobacteria) and malignancies such as Kaposi’s sarcoma and lymphomas (Mawle and Douglas 1992). In the absence of treatment, disease progression is usually relentless and almost invariably fatal.
Epidemiology of HIV Infection and Spread of AIDS
Since AIDS was first described in the US in 1981 in young, previously healthy, homosexual men, the disease has spread worldwide. An estimated 33.4 million individuals now live with HIV-1 infection and some 2.7 million are infected each year (Fauci and Folkers 2009). The disease is transmitted by sexual intercourse, sharing of needles, transfusion of blood and blood products and vertical transmission from mother to child. In the USA and Europe, sexual transmission by gay and bisexual men is still much more important than is heterosexual transmission, but the latter is increasing and appears to be the most important route of spread in Africa, Thailand, India and countries in South America and the Caribbean.
HIV-2 is more restricted in its geographical distribution and is concentrated mainly in West Africa, from the Cape Verde Islands to Benin (e.g. Gambia, Guinea Bissau) with limited spread to those countries in Western Europe historically associated with West Africa (Fleming 1990). Cases of HIV-2 infection have also been reported in East Africa, Asia, Latin America and North America. In several West African countries, the frequency of HIV-1 and HIV-2 infections is very similar. In children, AIDS caused by HIV-2 is more likely to be due to transmission by blood transfusion than perinatal transmission (Poulsen et al. 1989). Nevertheless, both serotypes have the same modes of transmission and AIDS caused by either is indistinguishable. Both HIV-1 and HIV-2 must be excluded from blood donations.
Transmission of HIV by Blood Transfusion
Recognition of a possible relationship between blood transfusions and the acquired immune deficiency syndrome (AIDS) provided early clues that AIDS might have an infectious cause. In July 1982, three patients with severe classic haemophilia who developed Pneumocystis carinii pneumonia were reported by the Centers for Disease Control in the Morbidity and Mortality Weekly Report (MMWR) (CDC 1982). All had been treated with large doses of commercially prepared factor VIII concentrate. Laboratory and clinical studies of these patients revealed evidence of impaired cell-mediated immunity. In quick succession, several additional reports linked haemophilia and AIDS, two relatively uncommon disorders (Ragni et al. 1983; White and Lesesne 1983). The association seemed unlikely to have happened by chance alone. In subsequent studies, some apparently healthy patients with haemophilia, including those treated only with cryoprecipitate obtained from volunteer blood donors, demonstrated abnormalities in cell-mediated immunity (Lederman et al. 1983).
The first reported case of transfusion-associated AIDS (TAA) turned out to be an 18-month-old infant with severe combined immunodeficiency who had been transfused repeatedly at birth and had received a unit of platelets from a donor who subsequently developed AIDS (Amman et al. 1983). By 1984, 38 cases of AIDS had been reported in patients with no risk factors other than a history of transfusion. Nineteen of the patients were adults who, during the previous 5 years, had received blood components derived from unpooled donations. In those cases in which all the donors could be followed up, an individual in a ‘high-risk’ group could always be identified (Curran et al. 1984). An expanded study of 194 patients showed that in most cases the high-risk donor was anti-HIV positive, and in those few cases in which the high-risk donor lacked anti-HIV, another donor tested positive (Peterman et al. 1985). A total of 157 525 cases of AIDS was reported in the USA by the end of 1990, and 5371 (3.4%) of these were attributed to the transfusion of blood or blood components. The median incubation period, estimated as the time from exposure by transfusion to diagnosis of AIDS, has been estimated to be 34 months in adults and 22 months in children (Peterman 1987), although later estimates indicated a longer period with a median of 7–8 years for adults and 3–5 years for children (Rogers and Schochetman 1992). In total, 50% of untreated subjects infected with HIV by transfusion will develop AIDS within 7 years compared with 33% of subjects infected by other routes (Ward et al. 1989).
Following the first reports, the number of cases of TAA in the USA increased rapidly. By the end of 1991, out of a total of 206 392 cases of AIDS, there were 6060 adults and 472 infants or children who had acquired the disease by transfusion of blood or blood products, representing 3% and 13.6% of the total cases in adults and children respectively (CDC 1992). The risk of transmission of HIV by blood transfusion is now trivial in countries in which donor education, encouragement of self-exclusion and screening for HIV by NAT and serology have been introduced. (Zhou et al. 2010) In these countries, the risk of transmission of HIV is almost solely attributable to donations given during the window period (see below).
Because HIV is both cell associated and present in plasma, all blood components are potentially infectious (Curran et al. 1985). The viral load has been estimated at between 1.5 × 104 tissue culture infective doses in a 250-ml unit of blood from an asymptomatic donor, to 1.75 × 106 from a unit drawn from a symptomatic person (Ho et al. 1989). The relative importance of the viral strain, concurrent infection by other blood-borne agents, cellular receptors for HIV and other genetic and acquired host factors, both for infection and for clinical course of HIV in transfusion recipients, has received a great deal of attention. (Vicenzi et al. 1997; Keoshkerian et al. 2003; Zaunders et al. 2004).
Follow-up studies have shown that 90–95% of patients receiving blood or blood components from anti-HIV-positive donors have become infected (Ward et al. 1987; Donegan et al. 1990b). The virus is well preserved in refrigerated and frozen blood; however, components that are washed, leucoreduced or cold stored for several weeks, procedures that diminish the number of viable leucocytes or the amount of virus, reduce the likelihood of transfusion transmission (Donegan et al. 1990a). Neither donor status nor recipient characteristics affect the likelihood of HIV transmission (Busch et al. 1990a). However, when AIDS developed in the donor shortly after donation, the period of asymptomatic infection in the recipient was also shortened (Ward et al. 1989). Albumin preparations, immunoglobulins, antithrombin III and plasma-derived hepatitis B vaccine have not been associated with HIV infection (Desmyter 1986; Melbye 1986; Morgenthaler 1989; Cuthbertson 1991). Furthermore, when HIV is added to plasma and the plasma is then fractionated by the cold-ethanol process, HIV does not appear in the Ig fraction (Morgenthaler 1989). Plasma for fractionation is both screened and treated to inactivate HIV, and no transmissions have been documented in appropriately manufactured plasma products in 25 years.
TAA in infants, and AIDS in infants and children in general, has a shorter incubation period than in adults and many of the clinical manifestations of the disease are different (Rogers and Schochetman 1992). Infants born to anti-HIV-positive mothers who become infected perinatally and infants transfused during the first years of life have the shortest median incubation period (less than 5 years), and usually develop AIDS in the first year of life. The increased susceptibility of infants to AIDS may be related to their immature immune system and to the larger viral load relative to body size. Children likely to develop TAA are those who are likely to be transfused: premature infants and children with haemophilia, thalassaemia and sickle cell anaemia. In the USA, of 2734 children with AIDS, 250 (9.1%) were transfusion associated and 138 (5.0% of the total) occurred in children with coagulation factor deficiencies (CDC 1991).
In much of Asia and Africa, the transmission of HIV by blood transfusion is still an important source of infection. Reasons for an alarmingly high rate of transmission, reported to be up to 10% of all cases, include: (1) the high demands for blood for inpatients with severe anaemia and haemorrhage, mainly in obstetrics, gynaecology and paediatrics; (2) the prevalence of HIV infection amongst the donor population, which can be as high as 20%; (3) the fact that HIV infection is not confined to a minority of the population who can be requested to refrain from blood donation; and (4) the inability of many laboratories to test for HIV or to perform and control the tests properly. The patient groups at greatest risk of acquiring HIV-1 or -2 by blood transfusion in tropical Africa are children with malaria and anaemia, patients with sickle cell anaemia (120 000 infants with sickle cell disease are born each year in Africa), anaemic antenatal patients, women with severe obstetric haemorrhage and trauma victims (Fleming 1990).
Transfusion-Associated AIDS and Haemophilia
To December 1996, 4674 cases of AIDS were reported in US patients with haemophilia, accounting for less than 1% of the 581 429 AIDS cases reported in adults and children. Of the 7629 cases of AIDS reported in children under the age of 13 years, 373 (5%) were recipients of blood or tissue and 231 (3%) were haemophiliacs (Centers for Disease Control and Prevention 1997). All but 39 of these infections occurred prior to testing blood and plasma donors for HIV.
Studies of patient cohorts and specimens in serum repositories revealed that more than 90% of severe haemophiliacs (subjects with less than 1% factor VIII activity) treated with factor VIII concentrate had been infected prior to 1984 (Evatt et al. 1985). In a study of a 16-centre cohort of haemophiliacs in the US and Europe, infections first appeared in 1978, peaked in October 1982 and declined to an estimated four infections per 100 person-years by July 1984 (Kroner et al. 1994). For patients who were high-dose recipients, peak risk appeared even earlier, indicating that the majority of patients with haemophilia were infected before the disease was widely recognized and long before it was attributed to transfusion. The risk was related to each patient’s mean annual dose of clotting factor concentrate. As clotting factor concentrates are prescribed on the basis of patient weight or plasma volume, older patients with severe disease generally received more concentrate, had more ‘donor exposures’ and seroconverted sooner than did children and patients with milder disease. When corrected for dose and severity of disease, the association between age and early seroconversion disappears. The cumulative incidence of infection was 96% for high-dose recipients, 92% for moderate-dose recipients and 56% for low-dose recipients. Subjects who received only single-donor products (plasma and/or cryoprecipitate) had the lowest cumulative incidence of infection, 16% (Kroner et al. 1994). This experience is consistent with other reports (Andes et al. 1988; Gjerset et al. 1991). These startling numbers underscore the potential public health risks of using transfusion products manufactured from pools of plasma drawn from 20 000 donors or more.
Although AIDS was first reported in three haemophiliacs in 1982, studies of serum samples stored from as far back as 1968 have shown that the first cases of the development of anti-HIV in haemophiliacs occurred in 1978 in the US and in 1981 in the UK (Evatt et al. 1983; Machin et al. 1985; Ragni et al. 1986). Plasma for preparing the implicated clotting factor concentrates may have been collected a year prior to that. The prevalence of HIV seropositivity and of AIDS varies from one haemophilia centre to another depending on the source, volume and type of concentrate used. In a study of 13 haemophilia centres in Western Europe, Canada and Australia involving 2370 patients with haemophilia A and 434 patients with haemophilia B, the overall incidence of antiHIV was 53.6% (CDC 1987a). The percentage of haemophiliacs infected with HIV in different countries has varied from 4% to more than 60%, with the higher rates in those countries using mainly concentrates from plasma imported from the US. Some batches of factor VIII concentrate from European plasma have also transmitted HIV (Madhok and Forbes 1990). A clear correlation was also found between the severity of haemophilia and HIV seropositivity (Melbye 1986; UK Haemophilia Centre 1986). Haemophiliacs treated before 1985 with cryoprecipitate alone have shown a low risk of HIV infection (Ragni et al. 1986; UK Haemophilia Centre 1986).
Patients with haemophilia B fared somewhat better (Evatt et al. 1984; Mannucci et al. 1985; Ragni et al. 1986; UK Haemophilia Centre 1986). The difference may be due partly to the uneven partitioning of HIV in infected plasma during fractionation, with HIV separating preferentially in the cryoprecipitate fraction (Aronson 1979; Morgenthaler 1989; Madhok and Forbes 1990). The source of plasma used for fractionation is also partly responsible for this difference. In many countries all the factor IX is prepared locally, whereas at least part of the factor VIII is imported from the US. Approximately 70% of patients in the USA with haemophilia A and 35% with haemophilia B developed HIV antibodies before the introduction of methods for viral inactivation in blood products (CDC 1987b). In a multicentre study of haemophiliacs treated in the UK between 1980 and 1984, 896 (44%) of 2025 patients with haemophilia A were positive for anti-HIV; 20 (6%) of 324 patients with haemophilia B and 11 (5%) of 215 patients with von Willebrand’s disease were seropositive.
There has been no transmission of HBV, HCVor HIV by US-licensed plasma derivatives since the introduction of effective virus inactivation procedures (Tabor 1999).
Prevention of Transfusion-Transmitted HIV Infection
Reducing the Residual Risk
The reduction in risk of transfusion-transmitted HIV over the past 25 years has been dramatic and reassuring. Nevertheless, enormous public concern persists. As in almost no other area, blood safety, and specifically the possibility of HIV transmission, provokes emotions and measures to further reduce risks that defy the conventional cost–benefit calculus. Actions to reduced residual risk fall into three general categories: (1) measures that can be introduced by blood collection facilities, such as improved donor screening, testing, education and exclusion techniques; (2) enlightened transfusion practice, such as judicious use of allogeneic blood components, appropriate use of autologous blood, and alternatives to transfusion (see Chapter 17); and (3) measures that depend on the development of new technologies, such as viral inactivation of cellular components and safe substitutes for blood.
Donor demographics have proved effective at identifying and excluding donors at high risk for infection and transmission of HIV (Busch et al. 1991a). Donors with high-risk profiles include men who have had sexual contact with other men since 1977, intravenous drug users, residents of high prevalence regions, prisoners, prostitutes, haemophiliacs who have received ‘unsafe’ clotting factor concentrates and the sexual partners of people in all of these groups. The year 1977 was chosen as the point of reference, because the first clinical cases of AIDS in the US were diagnosed retrospectively as far back as 1978 (Jaffe et al. 1985). The rate of seropositivity has been higher in paid plasma donors than in volunteers although current figures are hard to come by.
Measures to exclude such subjects from blood donor rolls are among the most important means of preventing the spread of AIDS by transfusion. Nevertheless, predonation medical screening and donor education are not infallible (Leitman et al. 1989). Between 1% and 2% of donors do not report risks that would disqualify them from blood donation, and donation incentives such as complimentary laboratory testing increases this rate (Glynn et al. 2001). In a study of blood donors found positive for hepatitis C antibody, 42% admitted to intravenous drug use on subsequent questioning, despite denying such use on predonation screening (Conry-Cantilena et al. 1996). In an anonymous mail survey of 50 162 volunteer allogeneic blood donors, 1.9% of the 34 726 respondents reported one or more risk factors that should have led to their deferral at the time of their last donation (Williams et al. 1997). Refinements in blood donor screening techniques, such as the use of illustrated risk-related activities, expansion of the screening questionnaire and interactive computer-based screening, have been introduced, but data supporting the effectiveness of such measures are lacking (Mayo et al. 1991). The reasons why some volunteer blood donors appear to disregard certain screening criteria are unknown and may have more to do with donor psychology than with inadequate screening and education.
Donors who are confirmed positive for HIV should be counselled and referred to specialized centres for follow-up. Appropriate interventions will not only help the donor to obtain long-term supportive care and to prevent further spread, but will also aid transfusion services to understand which groups of the population are HIV seropositive and still come forward to donate (Lefrere et al. 1992). Donor education and selection methods can then be modified accordingly.
Self-Exclusion (‘Self-Deferral’) of Donors
Retrospective analysis indicates that the risk of contracting AIDS from the transfusion of blood and blood components (other than products of plasma fractionation) prior to focused screening and testing was far higher than the one infection per million units transfused that had been estimated during the 1983–1985 interval. The risk of HIV transmission from transfusion in San Francisco has been calculated from its first appearance in 1978 to rise exponentially to a peak risk of approximately 1.1% per unit transfused in 1983 (Busch et al. 1991a). A retrospective study of heavily transfused patients with leukaemia in New York City revealed an overall risk between 0.02% and 0.11% per component transfused (Minamoto et al. 1988). The major blood collectors published a Joint Statement of Recommendations in January 1983 (American Association of Blood Banks et al. 1983), and the Public Health Service published recommendations in March 1983 (CDC 1983) that proposed such measures as public education, self-deferral for donors engaging in high-risk activity and confidential unit exclusion procedures (Pindyck et al. 1984, 1985). These measures proved unusually effective. An estimated 90% of men in high-risk categories self deferred. By 1984, the risk in San Francisco had dropped to less than 0.2% per unit. Screening with anti-HIV-1 in 1985 reduced the risk to about 1 in 40 000 units (Busch et al. 1991a).
Routine Screening Tests for HIV in Blood Donors
In most countries, screening tests for anti-HIV by the enzyme immunoassay (EIA) format are now compulsory for all blood donations. If reactive (‘repeat reactive’), additional tests for HIV using independent methods are used to confirm the diagnosis of HIV infection (Stramer et al. 2004). Early ELISA assays using disrupted purified virus were plagued by false-positive reactions. Current generation screening assays, using recombinant and synthetic antigens, have reduced false reactives dramatically while increasing test sensitivity for both the predominant and the variant viral strains (Busch and Alter 1995). Confirmatory strategies include use of the Western blot, notoriously subjective and complicated by non-viral bands (Kleinman et al. 1998), or use of a second EIA or a NAT assay of HIV RNA.
Approximately 66 million donations were screened by the American National Red Cross from September 1999 to 30 June 1998, resulting in 32 HIV NAT yield units (yield of 1 per 2 million) (Zhou et al. 2010). Most NAT yield donors were male and in their early 30s.
Although some antibodies to core and other antigens of HIV-1 and HIV-2 crossreact (Sazama et al. 1992), currently available tests are designed to detect both anti-HIV-1 and anti-HIV-2. Such combined assays are available as antiglobulin ELISAs and as sandwich ELISAs and are routinely used in several countries. Seroconversion in infection with HIV-1 is detected earlier in these combined assays than by anti-HIV-1 assays (Gallarda et al. 1994; Fiebig et al. 2003). Most of the combined assays have been found to be less sensitive for the detection of anti-HIV-2 than most anti-HIV-2 specific tests (Christiansen et al. 1996).
Some samples with anti-HIV are repeatedly positive in some, but negative in other screening assays. If only one screening test is used, such samples may give false-negative results (Hancock et al. 1993). One cause of such false negatives is that antibodies against subtype O, a variant found predominantly in West Africa, are not recognized in all screening assays (Loussert-Ajaka et al. 1994; Schable et al. 1994). At present group O prevalence is low in the USA and in Western Europe (Sullivan et al. 2000). False-negative reactions have also been found to be due to contamination with glove powder, inhibition by serum proteins, haemoglobin and certain anticoagulants (Sazama 1995).
Currently available ELISAs for HIV-1 antibodies detect HIV-1 subtype group 0. Soon after the discovery of an assay for HIV-Ab, transmission of HIV by blood from seronegative donors had been recognized (Esteban et al. 1985; Raevsky et al. 1986; Ward et al. 1988; Cohen and Munoz 1989). Studies reported detection of HIV-1 p24 antigen in analyses of stored blood specimens from plasma donors as early as 1986, and confirmed cases of HIV-Ag-positive, Ab-negative blood in primary HIV infection were reported by 1988 (Allain et al. 1986; Clark et al. 1991; Irani et al. 1991). The utility of this test as a screening assay was not so obvious. Antigen tests are positive for only part of the initial antibody-negative viraemic phase. In some subjects antigen can be detected as early as 2 weeks after infection, persisting for between 3 weeks and 3 months, and is no longer detectable when anti-p24 appears in the serum, although it may reappear intermittently during the asymptomatic phase (Allain et al. 1986; Fiebig et al. 2003). Later, antigen may reappear with a loss of anti-p24. A prospective study of 515 494 units donated at 13 blood centres in the US failed to detect a single instance of Ag-positive/Ab-negative donated blood. A retrospective analysis of 200 000 repository specimens and prospective studies of blood donors in Europe confirmed these findings (Backer et al. 1987; Busch et al. 1990a). However, after three anti-HIV seroconversions followed transfusion of p24 antigen-positive units, testing of donated blood for p24 antigen was mandated in the US in 1996 (Busch and Alter 1995). Mathematical models predicted that universal antigen screening would detect eight additional potentially infectious units per year. In fact it took 5 years before eight antigen-positive/antibody-negative units were interdicted (Kleinman et al. 1997; Kleinman and Busch 2000). Because of this limited usefulness and troubling frequency of false positives, HIV p24 antigen screening was not adopted widely outside of the US. With the adoption of universal NAT for HIV in the USA in 1999 and its licensure in 2002, HIV-Ag screening was rendered unnecessary (Busch et al. 2000).
Confirmatory Tests: They Do Not Always Confirm
The Western blot has been the most widely used additional or ‘confirmatory’ test for HIV. The criteria for the interpretation of Western blot results have been re-evaluated several times because of greater sensitivity and specificity of screening assays and Western blot reagents, better insight into the serological patterns of HIV infection, experience with patterns of non-specific reactivity in low- and high-risk populations and knowledge of the serological basis of non-specificity (Sayre et al. 1996). Samples are now considered to be WB positive demonstrate reaction with the gp41 and gp120/160 env bands or with either of these bands and the p24 gag band. The earlier requirement for a reaction with a third gene product (e.g. p31 or p66 pol bands) has been abandoned and reactivity with more than one env antigen alone is enough for confirmation (O’Gorman et al. 1991). If there is reactivity with only one band, the result of the Western blot is considered to be indeterminate. The absence of reactivity in the Western blot indicates that the donor has not developed anti-HIV (Dodd 1991).
In the original Western blot assay, viral lysate was used as a source of antigen. In the newer assays, recombinant or synthetic viral antigens are applied. These assays have been found to be both more sensitive and specific than the original WB (Soriano et al. 1994). Nevertheless, the assay is still relatively subjective and beset by indeterminate and false-positive results when compared with NAT as the ‘gold standard’ or when investigated with sequential sampling follow-up (Kleinman et al. 1998; Mahe et al. 2002; Stramer 2004). National programs are increasingly abandoning the Western blot in favour of algorithms that ‘confirm’ with different EIA assays and NAT.
Detection of HIV DNA and HIV RNA
Donations during the window period constitute the predominant risk for HIV transmission through transfusion (Busch et al. 2000). A more sensitive and specific alternative to testing for p24 antigen is NAT for HIV DNA in PBMC (Ou et al. 1988) or HIV-1 RNA in plasma by reverse transcription PCR or a similar amplification assay (Henrard et al. 1992). All blood in the US, Japan and most European countries is tested by NAT, often in pools of 16–90 specimens. Ultrasensitive assays can detect fewer than 10 genomic copies/ml (Busch et al. 2000). However, tests have been optimized for HIV subtype B and may lack sensitivity when applied to non-B subtypes (Triques et al. 1999). Single-donor combination assays (HIV/HCV/HBV) are in use in several countries.
Current Risk of Transmission of HIV by Blood Transfusion
Although the various measures outlined above have dramatically reduced the risk of TAA, a small residual risk remains (Delwart et al. 2004; Phelps et al. 2004). Most of this risk results from ‘window period’ donations. Before the introduction of HIV-Ag and NAT, prospective studies estimated this risk at about one infection in 60 000 units (Busch et al. 1991b; Nelson et al. 1992). Subsequent estimates rely on models based on calculations of HIV incidence and window period. In the US, residual risk has been calculated as 1 per 2 135 000 repeat donors (Dodd et al. 2002; Zou et al. 2012). The incidence rate is approximately two times greater among first-time donors. In most European countries where the prevalence of HIV in blood donors is lower than in the US, the residual risk is probably lower still. However, in countries with a high percentage of infected subjects and where HIV is spread mainly by heterosexual intercourse, the risk of transmission of HIV by blood transfusion is still considerable.
Human T-Cell Leukaemia Viruses Types I and II
The human T-cell leukaemia virus type I (HTLV-I), the first human retrovirus to be described, was isolated from cultured cells from a patient with an aggressive variant of Mycosis fungoides and from a patient with Sézary syndrome (Poiesz et al. 1980; Gallo et al. 1981). The virus has subsequently been shown to be identical to the adult T-cell leukaemia virus (Yoshida et al. 1982; Watanabe et al. 1984). HTLV-I is the causative agent of adult T-cell leukaemia (ATL) and is associated with a chronic demyelinating neurological disease called tropical spastic paraperesis (TSP), known in Japan as HTLV-associated myelopathy (HAM) (Vernant et al. 1987; Roman and Osame 1988). HTLV seropositive individuals appear to have a 0.25% lifetime risk of developing TSP, compared with a 2–5% risk of developing ATL (Kaplan et al. 1990). The virus is also associated with lung infections, cancer of other organs, monoclonal gammopathy, renal failure, infection with Strongyloides stercoralis, intractable non-specific dermatomycosis, lymphadenitis, periodontal disease, arthropathy, sicca syndrome, neurologic deficits, and uveitis. These effects may be due to the immunodeficiency induced by HTLV-I infection (Takatsuki 1996). The association of HTLV-I with Mycosis fungoides is controversial, as no HTLV-related DNA sequences could be detected in patients with this disease (Bazarbachi et al. 1993; Vallejo et al. 1995). In Japan, only 2.5% of HTLV-I carriers develop ATL (Takatsuki 1996).
HTLV-I and -II belong to the oncovirus subtype of the retrovirus family and are able to induce polyclonal proliferation of T lymphocytes in vitro and in vivo. Like the lentiviruses HIV-1 and -2, these viruses are lymphotropic and neurotropic, and have the essential structural genes gag (group antigen), pol (reverse transcriptase) and env (envelope) in addition to regulatory genes. In HTLV-I, the gag gene codes for the structural proteins p55/24/19; pol codes for a protein of approximately 100 kDa, and env codes for glycoproteins gp61/46/21. In HTLV-II the structural proteins are similar to those in HTLV-I with a high degree of crossreactivity; gag encodes the polypeptides p53/24/19; pol a protein of approximately 100 kDa; and env codes for the glycoproteins gp61/46/21. HTLV-3 and HTLV-4 have been found in subjects in Cameroon, but as yet there is no evidence of wider dissemination or disease association.
Areas endemic for HTLV have been found, particularly in southwest Japan, with prevalence as high as 15% (Maeda et al. 1984), in the Caribbean with a 1–8% prevalence (Clark et al. 1985), in regions of Central and South America and in parts of sub-Saharan Africa (Gessain et al. 1986; Vrielink and Reesink 2004). Populations in these areas show different prevalence rates for anti-HTLV-I, as do emigrants from these regions (Sandler et al. 1991; Vrielink and Reesink 2004). It has been estimated that more than 1 million Japanese people are healthy carriers of HTLV-I. Carriers of HTLV have also been found in the US, especially in Florida and states on the Pacific, and in France, UK, the Netherlands and many other countries. The prevalence in blood donors has been reported to be one in 6250 in the US (CDC 1990), about one in 30 000 in France (Pillonel et al. 1996), one in 45 000 in the Netherlands (Zaaijer et al. 1994) and one in 20 000 in London (Brennan et al. 1992).
HTLV-II, the second human retrovirus to be discovered, has a 65% nucleotide sequence identity with HTLV-I and a significant serological crossreactivity (Hjelle 1991). However, HTLV-II antibodies are not detected by all HTLV-I assays. The distinction between HTLV-I and -II can be made by DNA PCR (Reesink et al. 1994), and in the recently developed WB assays in which specific HTLV-I and -II recombinant antigens are used. The relative prevalence of the two viruses in blood donors in the US was found to be about equal (Glynn et al. 2000), but in many other countries HTLV-I predominates in blood donors. There is a high prevalence of HTLV-II among i.v. drug users and their sexual contacts in the US and other countries (Vrielink and Reesink 2004). A large proportion of HTLV-II-positive subjects in the US were Hispanic and American Indians (Hjelle et al. 1990a; Sandler et al. 1991). HTLV-II has been found to be associated with a HAM-like neurological disease (Hjelle et al. 1992; Murphy et al. 1997). Although the virus was first found in a patient with hairy cell leukaemia (Kalyanaraman et al. 1982) and subsequently in other T-cell malignancies, no viral RNA could be detected in the malignant cells (Manns and Blattner 1991). Both agents have been associated with pulmonary and urinary tract infections and autoimmune disorders (Murphy et al. 2004). In a prospective cohort of 155 HTLV-I, 387 HTLV-II, and 799 seronegative subjects, HTLV-II, but not HTLV I- status was strongly associated with increased all-cause and cancer mortality (Biswas et al. 2010).
Transmission of Human T-Cell Leukaemia Virus
HTLV is mainly transmitted by sexual contact, by the sharing of infected needles and from mother to child, particularly by breast-feeding (Kajiyama et al. 1986). If infected mothers refrain from breast-feeding, transmission of HTLV to their infants is prevented in 80% of cases (Hino et al. 1996). Transmission from mother to fetus has been demonstrated by culture studies of cord blood lymphocytes in 2 out of 40 cord blood samples from HTLV-I-positive mothers (Satow et al. 1991).
Human T-Cell Leukaemia Virus and Blood Transfusion
HTLV-I has been transmitted by cellular components, but not by cell-free plasma or plasma derivatives (Okochi 1985). However, HTLV-RNA is detectable in plasma from infected subjects. The lack of infectivity of HTLV-positive plasma may be explained by the presence of neutralizing antibodies, the fact that it is integrated in viral DNA and the requirement of cell–cell interactions for infectivity (Rios et al. 1996).
Antibodies are usually first detectable 14–30 days after transfusion, although the interval may be as long as 98 days (Inaba et al. 1989; Gout et al. 1990). Of 85 recipients of anti-HTLV-I-positive cell concentrates in Japan, 53 (62%) developed antibodies 3–6 weeks after transfusion: IgM antibodies were present only in the early stages, whereas IgG antibodies persisted at high titre throughout the period of follow-up (Sato and Okochi 1986). Storage of blood appears to decrease the risk of transmission of HTLV. This may explain the lower rate of transmission reported in US transfusion recipients, whose blood may have been stored for a longer period than the units in Japan and the Caribbean (Donegan et al. 1994). Antibodies became detectable in 79.2% of recipients of blood stored for 1–5 days, but in only 55% of recipients of blood stored 11–16 days (Okochi 1989).
Recipients of HTLV-I-infected concentrates may develop HAM (Gout et al. 1990; Araujo and Hall 2004). An estimated 2–8% of subjects infected with HTLV-I by blood transfusion will eventually develop HAM (Murphy et al. 1997; Araujo and Hall 2004). ATL developed in two immunosuppressed patients who had received multiple transfusions 6 and 11 years earlier (Chen et al. 1989). HTLV-II has also been transmitted by blood transfusion (Hjelle et al. 1990b).
Screening Tests for Human T-Cell Leukaemia Virus Antibodies
For the detection of HTLV antibodies, ELISAs are used as well as gelatin particle assays. In the ELISAs for the detection of anti-HTLV-I, viral lysate has been used. Although there is considerable (65%) crossreactivity between HTLV-I and -II, the sensitivity of anti-HTLV-I assays for detecting anti-HTLV-II was found to be only 55–91% (Wiktor et al. 1991; Cossen et al. 1992). As infection with HTLV-II is probably associated with HAM and as many HTLV-positive donors (more than 50% in the USA) are HTLV-II positive, tests designed to detect both antibodies are now used (US Food and Drug Administration 1998). In these ELISAs, recombinant proteins, including HTLV-I and -II-specific ones, have been added to lysate or are used exclusively (Hartley et al. 1991). The sensitivity of such assays has been claimed to be 100% (Vrielink et al. 1996a).
In Japan, an agglutination test in which gelatin particles are coated with HTLV-I-antigens has been developed and used extensively. The sensitivity of the commercially available agglutination test Serodia HTLV-I has been claimed to be 100% for detecting anti-HTLV-I; all of 12 anti-HTLV-II-containing sera gave positive reactions (Vrielink et al. 1996a). An ELISA for the combined detection of anti-HIV-1 and -2 and anti-HTLV-I and -II in which synthetic peptides of all four viruses are used, has been developed (McAlpine et al. 1992). In a study 242 samples from anti-HIV-1/-2 or HTLV-I/-II panels, two HTLV-II-positive samples and two very weak anti-HIV-1-positive samples were negative. The specificity of the test was slightly less than that of specific assays (Flanagan et al. 1995).
Despite the improved specificity of anti-HTLV-I/-II screening tests, many repeatedly positive reactions that cannot be confirmed are still found and all repeatedly reactive samples must therefore be tested in confirmatory assays. An HTLV-I/II chemiluminescent immunoassay (ChLIA; Abbott PRISM) assay has been used to screen donations in Australia for more than a decade and may be useful as a confirmatory assay (Stramer et al. 2011a).
Human T-Cell Leukaemia Virus Confirmatory Assays
Confirmatory testing for HTLV-I/-II continues to be challenging, primarily because of a dearth of licensed reagents. Western blot and radioimmunoprecipitation assays (RIPAs) are used (Anderson et al. 1989; Hartley et al. 1990). For a positive reaction, antibody reactivity with both a gag (p19 and/or p24) and an env protein (gp46 and/or gp68) are required (WHO 1990). All other reaction patterns were considered to be indeterminate. As the sensitivity of the Western blot in detecting antibodies against env proteins was low, many indeterminate results had to be checked in RIPA, a much more elaborate assay (Lillehoj et al. 1990; Lal et al. 1992). A report on the transmission of HTLV-I by blood from a donor with an indeterminate pattern in the WB and RIPA (p19 and gp68 reactivity) to four out of six recipients, confirmed by PCR, demonstrated the insufficient sensitivity of these original confirmation assays (Donegan et al. 1992). A modified WB has been developed in which both shared (r21e) and specific (rgp46I and rgp46II) HTLV-I and -II recombinant env proteins are used (Lillehoj et al. 1990; Lal et al. 1992). For a positive reaction in this modified Western blot, a reaction with at least one gag protein (p19 and/or p24) and with env r21e as well as rgp46I or rgp46II is required. All other patterns are indeterminate. This Western blot, in which a reaction with HTLV-I or -II can be distinguished, has been found to be more sensitive and specific than the original Western blot. Instead of 66, only two positive samples required confirmation by RIPA and none of 158 indeterminate samples (original Western blot) reacted (Brodine et al. 1993). A recombinant immunoblot assay (RIBA) in which the same antigens are applied gave similar results (Vrielink et al. 1996b). Neither NAT nor ChLIA has been licensed for confirmatory testing in the US.
Cytomegalovirus
Characteristics of the Virus and of Cytomegalovirus Infection
Cytomegalovirus (CMV) is a large, enveloped, double-stranded DNA, beta herpes virus that is cell associated, but may also be found free in plasma and other body fluids (Drew et al. 2003). CMV has a direct cytopathic effect on infected cells. The result may lead to neutropenia, some depression of cellular immunity and inversion of T-cell subset ratios, with a consequent increase in susceptibility to bacterial, fungal and protozoa infections in immunosuppressed patients (Grumet 1984; Landolfo et al. 2003). CMV infection causes parenchymal damage, such as retinitis, pneumonitis, gastroenteritis and encephalitis, and can result in substantial morbidity and mortality.
CMV can cause primary acute clinical and subclinical infections. Chronic subclinical infections may occur in which the virus is shed in saliva and urine. CMV remains latent in a large proportion of infected subjects. The presence of anti-CMV does not guarantee immunity. As with HIV and HCV infection, specific antibody is a marker of potential infectivity although only in the case of CMV, a relatively small proportion of seropositive subjects seem to be infectious (Drew et al. 2003). CMV antibody-positive subjects may infect others through sexual contact, breast-feeding, transplacental transmission or transfusion (Tegtmeier 1986). In subjects with antibody, CMV infection may be reactivated, or the subject may become re-infected with exogenous strains of CMV.
Primary CMV infection is generally more severe than is re-infection (co-infection with a different strain) or reactivation. In view of the difficulty of distinguishing between reactivation and re-infection, the term recurrent infection has been coined to embrace both. However, when necessary to distinguish between the two, donor viral DNA can be distinguished from recipient viral DNA by restriction enzyme analysis (Glazer et al. 1979; Chou 1990). In practice, the diagnosis of recurrent infection is limited to demonstrating a fourfold increase in antibody titre or the presence of IgM anti-CMV. In immunosuppressed patients, serological tests cannot be relied upon for a diagnosis of CMV partly because the patients may not make antibody and partly because any anti-CMV detected may have been derived from transfused blood. Viral culture is impractical because the virus grows slowly in vitro. On the other hand, immunofluorescence techniques to detect viral antigen using monoclonal antibodies on biopsies or bronchial washings provide results within hours (Griffiths 1984).
Prevalence of Anti-CMV
The frequency of subjects with anti-CMV varies widely in different populations. Seroprevalence is lower (30–80%) in developed than in developing countries, where the figure may reach 100% (Krech 1973; Preiksaitis 1991). The prevalence of anti-CMV correlates with age and socioeconomic status (Lamberson 1985; Tegtmeier 1986). The frequency of anti-CMV-positive donors may vary widely within a given country, for example 25% in southern California and 70% in Nashville, Tennessee (Grumet 1984; Tegtmeier 1986).
Transmission of Cytomegalovirus by Transfusion
The transmission of CMV by blood transfusion was first reported in the 1960s (Kaariainen et al. 1966; Paloheimo et al. 1968; Klemola et al. 1969). CMV is now known as one of the infectious agents most frequently transmitted by transfusion. The pathogenesis of transfusion-transmitted CMV infection is not clearly understood. In most cases CMV appears to be transmitted in a latent, particulate state only by cellular blood components, and the virus reactivates from donor leucocytes after transfusion. Host as well as donor factors are involved in CMV infection (Tegtmeier 1989; Preiksaitis 1991). CMV has been isolated from the mononuclear and polymorphonuclear cells of patients with acute infections. The specific cell type responsible for carrying the virus has not been identified, although mononuclear cells are the favourite candidates as hosts of CMV in latent infection. Fresh blood appears more likely than stored blood to transmit CMV infection, although no controlled studies document this impression (Tegtmeier 1986).
Some 3–12% of units have the potential to transmit CMV (Adler 1984), although most authors have reported a carrier rate of 1% or less (Tegtmeier 1986; Drew et al. 2003). The discrepancy may be related to the frequency of donor testing and the sensitivity and specificity of the assay used. Primary infection rates depend on the number of transfusions, age of blood, time of year and immunocompetence of the recipient (Tegtmeier 1989; Preiksaitis et al. 1988; Preiksaitis 2000). Donors with IgM anti-CMV appear to be more likely than others to transmit CMV (Lamberson et al. 1984). One large study found that only 0.5% of antibody-positive donors have detectable CMV DNA in their leucocytes (Roback et al. 2003).
At present, no rapid, easy way to identify infectious subjects exists. Viral excretion in urine is a good index of infectivity, but blood donors would probably rebel at this screening strategy. Virus can also be cultured from saliva, and PCR-based assays are available for the detection of viral genome in peripheral blood. Detection of pp65 antigen in leucocytes (pp65 antigenaemia) is considered the ‘gold standard’ among diagnostic tools for diagnosing CMV infection and initiating antiviral therapy. Both CMV DNA and immediate early-messenger RNA detection have been compared with pp65 antigenaemia, but none of them showed advantages in terms of earlier diagnosis and better prognosis. PCR’s major advantage is its semi-automation compared with the immunofluorescence employed for pp65 antigenaemia. Isolation of CMV by culture is reliable for the diagnosis of active infection, but is less sensitive and requires more time for viral detection.
Transfusion-Transmitted Cytomegalovirus Infection in Immunocompetent Subjects
Before the era of universal (or near universal) leucoreduction, some 30% of anti-CMV-negative recipients undergoing cardiac surgery involving transfusion developed infection, as confirmed by virus isolation or the development of anti-CMV. In addition, some anti-CMV-positive patients developed recurrent infection. In almost all cases, the infection is asymptomatic. Of patients who develop a primary or recurrent CMV infection following transfusion, fewer than 10% develop a mononucleosis-like syndrome. This syndrome, originally termed the post-perfusion syndrome, but now referred to as the post-transfusion syndrome, appears 3–6 weeks after transfusion. Common features include fever, exanthema, hepatosplenomegaly, enlargement of lymph nodes and the presence in peripheral blood of atypical lymphocytes resembling those found in infectious mononucleosis (Foster 1966). Recovery is usually complete. The development of atypical lymphocytes due to post-transfusion CMV infection should be distinguished from the development of atypical lymphocytes 1 week after transfusion as a response to allogeneic lymphocytes.
Consequences of Transfusion in Patients with Impaired Immunity
During the past two decades, major advances have been achieved regarding the management of CMV infection through the development of new diagnostic techniques for the detection of the virus and through the performance of prospective clinical trials of antiviral agents (Meijer et al. 2003). Nevertheless, in immunosuppressed patients, or in fetuses and premature infants with an immature immune system, CMV infections, particularly primary infections, still cause severe disease that can be fatal.
The Fetus in Utero
Following maternal primary CMV infection, the fetus becomes infected in 30–40% of cases. Approximately 5–10% of infected infants develop sequelae such as mental retardation, hearing loss or chorioretinitis (Stagno et al. 1986).
Premature Infants
The risk of serious CMV infection is high when the infant’s birthweight is less than about 1300 g and when the mother is anti-CMV negative. In two large prospective studies, 25–30% of infants with these risk factors, transfused with a total of 50 ml or more of blood, some of which was anti-CMV positive, acquired CMV infection, and 25% of these infants died. Infants transfused with anti-CMV-negative blood did not develop CMV infection (0 out of 90) (Yeager et al. 1981; Adler et al. 1983). A lower incidence of CMV infection (7–9%) has been reported from two other centres: in infants weighing less than 1500 g born to anti-CMV-negative mothers and transfused with blood, some of which was anti-CMV positive (Smith et al. 1984; Tegtmeier et al. 1984). All reports agree that clinically significant CMV infection in newborn infants develops only when the infant is premature and of low birthweight, when the mother lacks anti-CMV and when anti-CMV-positive blood is transfused (Tegtmeier 1986).
Bone marrow transplant recipients frequently develop primary or recurrent CMV infections that may prove fatal (Tegtmeier 1986). Blood transfusion represents the main risk factor for CMV acquisition in CMV-negative patients receiving bone marrow from a CMV-negative donor. In a prospective randomized trial of 97 anti-CMV-negative patients, 57 received anti-CMV-negative marrow: 32 out of the 57 received anti-CMV-negative blood components and only one of these developed a CMV infection; of the 25 who received blood components unscreened for anti-CMV, eight developed CMV infection. Among the 40 recipients of anti-CMV-positive bone marrow, the rate of CMV infection was no lower in those who received only anti-CMV-negative blood components (Bowden et al. 1986). Granulocyte concentrates, which contain large numbers of leucocytes that can harbour CMV, reportedly carry the greatest risk of transmitting CMV infection (Winston et al. 1980; Hersman et al. 1982). However, when only two prophylactic transfusions were given, the risk appeared to be no higher in those who received granulocytes than in those who did not (Vij et al. 2003).
Renal transplant recipients are at high risk of primary or recurrent CMV infection; the main source of infection lies in the transplanted kidney (Tegtmeier 1986). In anti-CMV-negative recipients of a kidney from an anti-CMV-negative donor, blood transfusion plays a significant role in CMV transmission (Rubin et al. 1985).
Heart and heart–lung transplant recipients may develop severe primary CMV infection, which can lead to opportunistic infections with fungi or bacteria. In anti-CMV-negative recipients the main sources of infection are anti-CMV-positive transplanted organs or organ blood donor units (Preiksaitis et al. 1983). If both donor and recipient are anti-CMV negative, blood transfusion is a major source of CMV disease; infection can be minimized by the use of anti-CMV-negative red cells and platelets (Freeman et al. 1990). If a heart or heart–lung from a donor with CMV antibodies is transplanted to an anti-CMV-negative recipient, prophylactic administration of specific CMV hyperimmune IVIG seems to lessen the severity of CMV disease (Freeman et al. 1990), although data in support of this practice are unimpressive.
Following splenectomy due to trauma, patients receiving massive transfusion may develop serious CMV infections (Baumgartner et al. 1982; Drew and Miner 1982).
Subjects infected with HIV and especially those with AIDS, if anti-CMV negative, may acquire primary CMV infection by transfusion (Jackson et al. 1988; Sayers et al. 1992). In these patients CMV may cause sight-threatening infection that may lead to blindness in up to 25% of patients not receiving antiviral therapy. Because the rate of reactivation of CMV in already infected patients is high, it is difficult to determine the contribution of transfusion-transmitted virus (Bowden 1995).
Liver transplant recipients should also be considered at risk, especially children or pregnant women. Now that smaller amounts of blood and blood components are needed in liver transplantation, it has been possible to give only anti-CMV-negative blood and platelets to anti-CMV-negative recipients of anti-CMV negative grafts.
Prevention of Transfusion-Transmitted Cytomegalovirus Infection
Subjects who are at highest risk of severe primary CMV infection are anti-CMV-negative patients with impaired immunity. For such patients, preventive measures are available to reduce transmission of CMV by transfusion. The selection of CMV-seronegative donors has proven to be effective, but not infallible (Tegtmeier 1989; Miller et al. 1991). Seropositivity, especially the presence of IgM, is a marker of previous infection and latent, but potentially infectious virus. However, antibody assays vary in sensitivity and a small risk of transmission even from seronegative units remains (Kraat et al. 1993; Bowden et al. 1995). Window period infections are the most likely source of antibody screening failures. Although the window periods for HIV-1, HCV and HBV have been reasonably well defined, the length of the CMV-seronegative window, estimated at 6–8 weeks, is less well characterized. Several recent studies using PCR technology documented CMV DNA in both plasma and cellular blood components from several weeks before seroconversion to several months after seroconversion, although culture positivity was found for a much shorter period in white blood cells (WBCs) and not observed in plasma (Zanghellini et al. 1999).
When only red cells are required, frozen deglycerolysed red cells can be used and have not been shown not to transmit CMV (Brady et al. 1984; Taylor et al. 1986; Sayers et al. 1992).
As CMV is a white cell-associated virus, an alternative approach to preventing CMV transmission involves filtration removal of leucocytes from red cell and platelet concentrates (Graan-Hentzen et al. 1989). Leucoreduction by filtration may fail to prevent CMV transmission, as 105 to 106 WBCs may still be transfused and an estimated 1 in 1000 to 1 in 10 000 WBCs are infected by CMV during latency (Drew et al. 2003). In 10 patients whose WBCs were CMV antigen and culture positive before filtration and culture negative afterwards, 2 out of the 10 did, however, have CMV DNA detected in leucocytes after filtration (Lipson et al. 2001). Plasma viraemia, if present, would not be diminished by leucoreduction and might also account for CMV transmission following leucoreduced components. Although the exact number of residual leucocytes that is sufficiently small to pose no risk of CMV transmission is unknown and may not exist, a large prospective study has shown that leucocyte removal is as safe as selection of anti-CMV-negative donors. In this study, 502 patients were randomized prior to bone marrow transplantation to receive filtered, three-log leucocyte-depleted cellular components or components from anti-CMV-negative donors. There was no significant difference in the probability of CMV infection between the recipients of anti-CMV-negative or filtered concentrates, although more CMV associated disease was observed in the filtration group (Bowden 1995). The same investigators have subsequently questioned their finding that filtered red cells are equivalent to CMV-seronegative cells (Nichols et al. 2003). Whereas leucodepleted components are commonly termed ‘CMV-safe’ as contrasted with ‘CMV-negative,’ no evidence indicates that one preparation is preferable. Many paediatricians prefer CMV seronegative components for their low birthweight premature neonates, especially in view of high mortality of infections in this population. Since blood in most developed countries is already leucoreduced, perhaps seronegativity adds an additional layer of safety; however a trial to demonstrate additional benefit has not been performed and is unlikely to be undertaken. When granulocyte transfusions are needed, selection of anti-CMV-negative donors is the only solution.
The success of leucocyte removal in the prevention of CMV transmission has raised the question of the importance of infection with a second strain of CMV in anti-CMV-positive recipients and the value of transfusing such recipients with leucocyte-depleted concentrates. Although co-infection with a second strain of CMV does occur (Boppana et al. 2001), the clinical consequences of such infection resulting from transfusion are less clear.
Tests for Anti-CMV
Several tests for anti-CMV are available. Complement fixation used to be the diagnostic reference test, but it proved too complicated for routine donor screening. Indirect immunofluorescence, solid-phase fluorescence, ELISA and particle agglutination assays are also available. Competitive ELISAs seem to be the most reliable of the currently available screening tests (Bowden et al. 1987). CMV PCR technology has been used in clinical diagnostics since the 1980s (Bowen et al. 1997; Lipson et al. 1998). Conventional PCR requires detection and confirmatory testing of the amplicon by gel electrophoresis, incorporating an isotopically labelled or non-radioactive hybridization assay or a nested PCR to detect targets present in very low copy numbers (Lipson et al. 1995). A simplified, more rapid PCR-solid-phase enzyme immunoassay (EIA) plate technology system has been developed (Davoli et al. 1999). The quantitative real-time CMV PCR assay using TaqMan chemistry and an automated sample preparation system has also been applied to CMV detection (Piiparinen et al. 2004).
An inexpensive, uncomplicated CMV-Ag (pp65 antigen) assay is available and well suited for most diagnostic microbiology laboratories (Lipson et al. 1998). Assay for the early antigen pp65 is considered the ‘gold standard’ for the initiation of antiviral therapy.
Other Viruses
Epstein–Barr Virus
Infection with Epstein–Barr virus (EBV), a herpes virus-like CMV, is endemic throughout the world. EBV can cause primary symptomatic infection (infectious mononucleosis), but most commonly causes asymptomatic infection followed by latent infection (Henle 1985). In most countries more than 90% of blood donors have neutralizing anti-EBV, which coexists with latent virus in B lymphocytes of peripheral blood and lymph nodes. At least one in 107 circulating lymphocytes of carriers harbour EBV genomes, but post-transfusion EBV infection is a rare occurrence, and symptomatic infection is even rarer (Rocchi et al. 1977). The virus is found in three of every 104 peripheral lymphocytes during acute infection. The majority of susceptible recipients are young children. Even in children, the chance of acquiring EBV infection following the transfusion of anti-EBV-positive blood is minimal, because the donor’s neutralizing antibodies persist in the recipient’s circulation long after the EBV-infected lymphocytes disappear. In a study of 25 EBV antibody-negative patients aged 3 months to 15 years and transfused with 1–11 units of blood stored for not more than 4 days, only one developed EBV antibodies and this patient had no symptoms (Henle 1985).
Of five patients transfused during cardiac bypass, four who were initially anti-EBV negative produced antibody that persisted at high titre for many months postoperatively. Two out of the four patients had concomitant CMV infection and no heterophile antibodies; one developed transient fever, and the other hepatitis. Only one of the four patients developed an infectious mononucleosis-like syndrome with heterophile antibodies (Gerber et al. 1969).
Although most cases of ‘post-transfusion syndrome’ are caused by CMV, two adult patients who were anti-EBV negative before transfusion developed post-transfusion syndrome due to EBV (McMonigal et al. 1983). In a survey of some 800 patients, fewer than 8% lacked EBV antibody before transfusion, and only 5% of these developed antibodies following transfusion. These patients suffered no clinical illness or disturbance of liver function (MRC 1974). The discrepancy between these observations and those of others may have been related to the use of fresh blood in the last-named series (Gerber et al. 1969).
Post-transfusion infectious mononucleosis is seen only rarely in anti-EBV-negative immunocompetent patients and usually occurs when only a single unit of blood or blood component, obtained from the donor during the incubation phase is given within 4 days of collection. When more than 1 unit is transfused, one of the units is almost certain to contain anti-EBV. In the reported cases, the donors developed symptoms of mononucleosis 2–17 days after blood donation. The incubation period in recipients has been 21–30 days (Solem and Jorgensen 1969; Turner et al. 1972). Transfusion-transmitted EBV infection with symptomatic mononucleosis has occasionally been reported in patients transfused for splenectomy (Henle 1985).
Post-transfusion EBV infections may contribute to the development of lymphomas in severely immunosuppressed patients such as haematopoietic graft recipients. T-lymphocyte suppression allows EBV-infected B lymphocytes to outlive the passively transfused antibodies and to proliferate (Marker et al. 1979; Hanto et al. 1983).
Other Herpes Viruses
Herpes simplex and Herpes varicella zoster have never been shown to be transmitted by blood transfusion; viraemia occurs only during primary infections, which usually occur in childhood (Henle 1985).
HHV-6 is a human herpes virus originally named HBLV (human B lymphotropic virus) for its ability to infect freshly isolated B cells. The virus was found in patients with various lymphoproliferative disorders. HHV-6 can infect monocytes, macrophages, T cells and megakaryocytes (Ablashi et al. 1987). The virus is cytopathic for selected T-cell lines. Infection is acquired usually within the first year of life and the virus was found to be ubiquitous in blood donors when tested in London and the US (Briggs et al. 1988; Saxinger et al. 1988). No transfusion-associated disease has been reported.
HHV-8, Kaposi’s sarcoma-herpes virus is a white cell associated virus that has infected humans for thousands of years and has been the implicated agent for Kaposi sarcoma, primary effusion lymphoma, and Castleman disease. Despite a relatively high HHV-8 seroprevalence in a Texas blood donor cohort (23%), HHV-8 DNA was not detected in any sample of donor whole blood using a highly sensitive PCR assay (Hudnall et al. 2003). Transfusion of fresh, leucoreplete blood in sub-Saharan Africa where HHV-8 prevalence approximates 40% provides strong evidence of transfusion transmission to a small minority of recipients, but little evidence of associated disease (Hladik et al. 2006). Passive antibody transfer is common. Blood that undergoes leucoreduction and extended refrigerated storage is even less likely to transmit infectious virus. Blood components from HHV 8-infected donors apparently carry little clinical risk for the transfusion recipient (Engels et al. 1999).
Human Parvovirus B19
HPV B19 infection has long been known to cause erythema infectiosum (Fifth disease), a common febrile exanthem of childhood (Anderson et al. 1983). HPV is also associated with polyarthritis and rash in adults as a result of antigen–antibody immune complex deposition in skin and joints (Reid et al. 1985; White et al. 1985). Because of its specific cytotoxic effect on erythroblasts, HPV B19 can precipitate aplastic crises in children who have haematological disorders with shortened red cell survival, such as those with sickle cell anaemia and other chronic haemolytic anaemias, particularly hereditary spherocytosis (Pattison et al. 1981; Young and Mortimer 1984). The virus may also cause thrombocytopenic purpura (Pattison 1987). Intrauterine infection may cause hydrops fetalis and spontaneous abortion in early pregnancy (Brown et al. 1984; Anand et al. 1987).
HPV B19 was discovered by an Australian virologist who noted viral particles in an antigen–antibody line of detection (plate B, well 19) in an assay for hepatitis B (Cossart et al. 1975). HPV B19 is a small single-stranded, non-enveloped, thermostable DNA member of the Parvoviridae family (Shade et al. 1986; Young and Brown 2004). The parvoviruses are dependent on help from host cells or other viruses to replicate. Parvovirus B19 is the type member of the erythrovirus genus, which propagates best in erythroid progenitor cells. The red cell P antigen, a globoside present on a variety of cells in addition to erythrocytes, has been documented as the specific receptor for HPV (Brown et al. 1994). This may account for reports of polyarthritis nephropathy, myocarditis and cardiomyopathy (Young and Brown 2004). Persistent infection with anaemia occurs in immunosuppressed subjects (Kurtzman et al. 1987). PCR assays have revealed the presence of viral DNA along with the simultaneous presence of specific IgG in 0.55–1.3% of normal blood donors (Candotti et al. 2004), but the long-term persistence, infectivity and clinical importance of the virus in these subjects has not been well studied. Current methods of viral inactivation may not be able to eliminate HPV B19 completely (Williams et al. 1990a).
Infection with the virus normally occurs via respiratory droplets. HPV B19 has been transmitted by plasma fractionation products derived from large pools of plasma, particularly by factor VIII and factor IX concentrates (Santagostino et al. 1997; Blumel et al. 2002). One episode of fulminant hepatitis has been attributed to the intravenous route of infection (Hayakawa et al. 2002). Use of recombinant products should eliminate this risk (Soucie et al. 2004). Transmission of the virus by single-donor components is distinctly unusual, but a red cell unit has been associated with HPV B19 transmission and possible cardiac involvement in a 22-year-old woman with thalassaemia major (Zanella et al. 1995). Transmission from single donor blood components, correlates with viral titer, rarely occurs with titres <106 IU/mL, and may be influenced by the titre of neutralizing antibody in donor or recipient (Kleinman et al. 2009a; Hourfar et al. 2011).
The classic diagnosis of infection with HPV B19 is based on the detection of IgM or IgG antibodies with an EIA (Cohen et al. 1983). Alternatively viral DNA can be detected by PCR (Salimans et al. 1989; McOmish et al. 1993). HPV B19 has been detected by PCR in solvent–detergent-treated clotting factor concentrates (Lefrere et al. 1994), in heat-treated factor VIII and IX concentrates and in IVIG (McOmish et al. 1993; Santagostino et al. 1994) and in clotting factor concentrates prepared by different purification and inactivation procedures (Zakrzewska et al. 1992; Blumel et al. 2002). The virus has also been detected in plasma pools designed for fractionation (in 64 out of 75 pools), in 3 out of 12 albumin preparations, in 3 out of 15 IVIG and in three out of four IMIG preparations, as well as in seven out of seven factor VIII preparations. There is some indication that treatment of IVIG preparations at low pH may result in removal of detectable HPV B19 DNA (Saldanha and Minor 1996). Since 2002, major plasma fractionators have screened plasma units with quantitative measurements of B19 DNA to reduce the risk of transmission. The prevalence of HPV B19 in blood donors has been estimated at 0.03% in the UK (McOmish et al. 1993). A similar prevalence was found in Japan (1 in 35 000) but, during an epidemic of erythema infectiosum, the prevalence was much higher (1 in 4000) (Tsujimura et al. 1995). B19 prevalence varies according to season and from year to year (Young and Brown 2004).
A rapid test for HPV B19, suitable for large-scale screening of donors has been developed (Sato et al. 1996). The test is based on agglutination of blood group P-positive gluteraldehyde-treated red cells by the virus for which the P antigen is the receptor (see Chapter 4). Although only intact viruses can bind to P, the test has been found to be sensitive and it could be used to select donors for patients at particular risk. Routine screening of donors seems unnecessary. Although 81.6% of 136 haemophiliacs studied had anti-HPV B19, B19 DNA was detectable in none and there were no signs of lasting clinical or haematological sequelae (Ragni et al. 1996).
Commercial immune globulins are a good source of antibodies against parvovirus; a persistent B19 infection responds to a 5- or 10-day course of immunoglobulin at a dose of 0.4 g per kilogram of body weight, with a prompt decline in serum viral DNA, as measured by hybridization methods, accompanied by reticulocytosis and increased haemoglobin levels (Kurtzman et al. 1987; Frickhofen et al. 1990).
Arboviruses: Mosquitoes, Ticks and the Vertebrate Host
More than 100 arthropod-borne viruses (arboviruses) cause human disease, however relatively few transfusion transmissions have been proven, probably because most occur in the setting of large, explosive, mosquito-spread epidemics. Arboviral disease incidence varies according to the climatactic conditions that influence the vector. Outbreak frequency appears to be increasing globally due to changes in environment and human travel and land use. Three examples of arboviruses associated with transmission are described below, but the potential risks of the numerous other agents have been described and modelled (Petersen and Busch 2010).
West Nile Virus
Since its importation into the US in 1999, West Nile virus (WNV) has become a significant transfusion-transmitted infection with a calculated mean risk of transfusion transmission of 3.02 per 10 000 donations in high-risk metropolitan areas during epidemic conditions (Biggerstaff and Petersen 2003). Transfusion transmission was first documented when four organs harvested from a common multi-transfused cadaver donor transmitted virus to all four recipients; the donor’s pretransfusion sample tested negative for WNV, while one of 63 blood donors tested positive with a nucleic acid assay and developed IgM antibody to WNV over the subsequent 2 months (Iwamoto et al. 2003). Using strict case definition criteria, epidemiologists document 23 transmissions and another 19 inconclusive investigations of 61 case investigations during a 12-month period (Pealer et al. 2003). The true number of transmissions was probably higher by at least an order of magnitude.
As was the case with HIV and the hepatitis viruses, blood transfusion of WNV represents a small, but highly visible portion of a large epidemic. WNV is a mosquito-borne flavivirus transmitted primarily to birds and some small mammals. Humans serve as an incidental host. Approximately 80% of human infections are asymptomatic; 20% result in a febrile illness known as West Nile fever. About 1 in 150 patients develop meningoencephalitis and residual neurological deficits have been reported. Although there is no particular susceptibility to mosquito-borne infection, elderly and immunosuppressed subjects appear particularly vulnerable to severe, progressive disease. The virus is present for a week or more during initial infection. Most symptomatic subjects describe fever, headache and malaise, although these are not sufficiently specific for effective donor screening. Viral shedding may persist for 7–8 weeks after infection, usually in the presence of specific antibody. The period of infectivity has not been well defined.
Of the 23 well-studied patients with transfusion-transmitted infection, 14 were identified because West Nile virus-associated illness came to the clinician’s attention following transfusion. Overall, 15 patients had recognized illness (13 meningoencephalitis, two fever). The illness began between 2 and 21 days after the implicated transfusion. The highly immunosuppressed transplant recipients appeared to have the longest incubation periods (median 13.5 days). Red cells, platelets and fresh-frozen plasma (FFP) have all been implicated in transmissions. Sixteen blood donors were implicated in transmission, with no predilection for age or gender. Nine out of the 16 recalled symptoms compatible with a viral illness around the time of donation, although all donors passed the donor screening procedures. Three donors developed symptoms prior to donation, one on the day of donation and five post donation. Fever, headache and weakness were the most common symptoms. Samples obtained at the time of donation had virus levels less than 80 pfu per millilitre and all were negative for West Nile virus IgM antibody.
Since screening of blood was undertaken in the US, several thousand viraemic blood donations have been removed from the blood supply. Nevertheless, cases of WNV transmitted by transfusion still occur because of transfusion of components containing low levels of virus not detected by the testing of pooled specimens (Macedo et al. 2004). All blood donations in the US are currently screened by rtPCR of pooled samples, and the appearance of a positive donor in a geographic region adjacent to an epidemic area triggers screening of individual donor units (Kleinman et al. 2009b).
Similar flaviviruses such as St Louis encephalitis virus, and Japanese encephalitis virus are likely to be blood transmissible as well, although documentation is lacking.
Dengue Virus
Dengue is the most common Arboviral disease in humans. As many as 90% of infections may be asymptomatic. Approximately 500 000 cases are reported to WHO each year; estimates suggest that some 50–100 million cases occur worldwide and result in thousands of deaths (Whitehorn and Farrar 2010).
Dengue virus (DENV) is another single-stranded RNA virus of the Flaviviridae family. Infection with each of the four serotypes (DENV-1,-2,-3,-4) confers lifelong serospecific immunity. DENV virus is transmitted by Aedes spp. mosquitoes. The most common vector in the Americas is Aedes aegypti. Dengue is a tropical disease found in Central and South America, Africa, Asia and parts of Australia, but also results in epidemics in the US in Puerto Rico, and has recently been reported in southern Florida and the Texas-Mexico border. The incubation period is approximately 5 days and viraemia is present at least a one day prior to the onset of symptoms. The viraemia persists for about a week.
The name ‘Dengue’ is thought to originate from the Spanish transliteration of the Swahili expression ‘ki denga pepo’, meaning ‘cramp-like seizure caused by the overtaking by an evil spirit’. The clinical disease ranges from mild fever to classic dengue ‘breakbone fever’ (DF) with headache, retro-orbital pain, myalgia, arthralgia, thrombocytopenia, and rash to dengue haemorrhagic fever (DHF), and dengue shock syndrome (DSS) (Castleberry and Mahon 2003). The uncomplicated acute febrile illness usually lasts for 5–8 days. Increased vascular permeability is seen in dengue and is associated with high levels of cytokines including tumour necrosis factor alpha (TNF-α), interferon gamma (IFN-γ), interleukin 6 (IL-6) and interleukin 2 (IL-2). The disease can evolve into DHF and DSS with frank haemorrhage, capillary leak syndrome and shock. Many of the clinical features of dengue infection may be due to the patient’s immune response. Treatment is supportive; there is no vaccine.
Like WNV, DENV can be transmitted by blood transfusion, bone marrow and organ transplantation, as well as by needlestick and transplacental passage. Reports of transfusion-associated disease are uncommon. A single case has been reported from Hong Kong where DENV is non-endemic, and a cluster of cases attributed to components from a single donation was reported from Singapore, an endemic area (Chuang et al. 2008; Tambyah et al. 2008). A survey of blood donors in Puerto Rico in 2005 revealed a prevalence of dengue viral RNA in blood donations = 0.73/1000. No information is available concerning the clinical outcome of the donors or recipients of virus-containing blood components (Mohammed et al. 2008).
Chikungunya Virus (CHIKV)
Chikungunya, which means ‘that which bends up’ or ‘to dry up or become contorted’ in the Makonde language, is a disease caused by a single-stranded RNA Alpha virus transmitted by Aedes mosquitoes; CHIKV evolves rapidly, and a recent mutation may have contributed to the first outbreak on the European mainland. CHIK is endemic in sub-Saharan Africa, southern India and Pakistan, Southeast Asia, Indonesia, and the Philippines. An outbreak in 2006 in India and islands in the Indian Ocean resulted in more than 1.25 million suspected cases and attack rates approaching 45%. On La Réunion Island, the epidemic peaked in February 2006 with 35% of the population experiencing some clinical form of the disease. At that point all blood collection was suspended and components were airlifted from France. During 2005–2006, cases of CHIK fever were confirmed in US travellers who visited endemic areas and cases were reported in Revenna, Italy, the first outbreak on the European mainland (Centers for Disease Control and Prevention 2006; Rezza et al. 2007).
Unlike WNV and dengue, most CHIKV infections appear to be symptomatic. Fever, chills, headache, and severe arthralgia, primarily affecting small joints, herald the onset, and as with other arboviruses, rash, photophobia, conjunctival injection, and abdominal pain are common. Symptoms are less severe in children. Joint pain may become chronic in adults. Acute illness usually lasts 5–7 days. Haemorrhage, meningoencephalitis and myocarditis are rare. Morbidity can be significant, but mortality due solely to CHIKV infection has not been confirmed. ELISA or immunoflourescence may be used to detect IgM after 5 days of onset. Reverse transcriptase PCR may detect virus in early infections. Commercial tests are not available in the US.
Simian Foamy Virus
Simian foamy virus (SFV) (spumaretrovirus) is a highly prevalent primate retrovirus that has been shown to infect human cells, replicate and produce cell-free infectious virus. Tropism is broad and includes B and T lymphocytes, macrophages, fibroblasts, endothelial cells and kidney cells. Simian retroviruses are spread in areas of the world such as Central Africa, where non-human primates are hunted for food and among people who handle non-human primates as pets or laboratory animals. In total, 1% of people living in southern Cameroon were found to harbour antibodies to SFV; all had reported exposure to fresh primate blood or body fluids (Wolfe et al. 2004).
Studies from the US CDC report a prevalence of infection of 2–5% with simian foamy viruses among laboratory and zoo workers occupationally exposed to non-human primates (Heneine et al. 1998; Switzer et al. 2004). Persistent viraemia has been detected in peripheral blood lymphocytes (PBLs) and 11 SFV-infected workers are known to have donated blood. Six donors were confirmed positive at the time of donation. SFV transmission through transfusion was not identified among four recipients of cellular blood components (two RBC, one filtered RBC, one platelet concentrate) from one SFV-infected donor (Boneva et al. 2002). Derivatives containing plasma from that donor tested negative for SFV.
Evidence of SFV infection included seropositivity, proviral DNA detection and isolation of foamy virus. There is no evidence as yet that SFV causes human disease but, recombination within the host, especially in immunocompromised hosts that may allow persistent infection, remains a concern.
Prion Proteins
A fundamental feature of prion diseases involves a normal protein constituent of human tissue, prion protein (PrP) (Bendheim et al. 1992). PrP can exist in either a natural cellular form (PrPC) or as variant pathological forms known collectively as ‘scrapie isoforms’ or PrPSc. Different prion strains result in distinct clinical and pathological diseases (Prusiner 1998). Both forms of prion protein have an identical amino acid sequence, but differ in the secondary structure, beta-pleated structures in the variant forms instead of the alpha-helical structure of the native protein (Pan et al. 1993). The structural difference has two major consequences. First, the beta-pleated conformation allows PrPSc molecules to form protease-resistant aggregates. Second, the abnormal protein seems to be capable of suborning normal protein to the abnormal form. The molecules with beta-pleated regions cause key alpha-helical regions of the native protein to assume the pleated conformation, thus converting normal protein to abnormal aggregates that cause the spongiform degeneration of the brain that is characteristic of these diseases.
Prion proteins are linked to the cell plasma membrane by the lipid glycophosphatidyl inositol (GPI). GPI-linked proteins are released from cells by several different mechanisms and may be taken up by other cells, thus facilitating dissemination (Devetten et al. 1997). Prion proteins are taken up selectively by motile follicular dendritic cells, suggesting that infective proteins may be present in circulating blood (Klein et al. 2001).
Creutzfeldt–Jakob Disease
Creutzfeldt–Jakob disease (CJD) is a rare, but invariably fatal neurodegenerative disease, with an incidence of about one per million of the UK and US population (Collinge and Rossor 1996). CJD may be ‘sporadic,’ arising from a random change in a normal individual, ‘familial,’ arising from a point mutation in the DNA coding for the protein, which leads to an increased susceptibility to production of the abnormal form, or ‘acquired,’ by the transfer of infectious material, as in individuals injected with pituitary-derived hormones or treated with dura mater transplants (Brown et al. 1992). A single case of transmission from a corneal transplant has also been reported. The proportion of recipients acquiring CJD from growth hormone varies from 0.3% to 4.4% in different countries, and acquisition from dura mater varies between 0.02% and 0.05% in Japan, where most cases occurred (Brown et al. 2000). Patient follow-up after point exposures to contaminated materials indicate that clinical latency for iatrogenic CJD may exceed 30 years (Fradkin et al. 1991; Brown et al. 2000).
Classic CJD typically affects older subjects with a median age of 60 years. Memory loss is an early manifestation, but the disease progresses rapidly after the first symptoms through confusion, motor and cerebellar symptoms and dementia. Death often occurs within a year of the first symptom.
Because of the possibility of transfer of infectious prions by transfusion, donors have been questioned to discover (1) whether there is any family history of CJD; (2) whether they have received pituitary-derived hormones; or (3) in some countries, whether they have received corneal grafts or grafts of dura mater. However, a systematic review of five case–control studies from the UK, Europe, Japan and Australia, involving 2479 patients, failed to find an association between CJD and blood recipients. No evidence of transmission of CJD by blood transfusion exists, despite the identification of individuals who were exposed to blood donated by people who later developed this disease (Wilson et al. 2000). A study of preserved brain samples of 25 haemophilic patients who have high exposure to blood transfusions and potentially higher exposure to blood infected with the agent responsible for Creutzfeldt–Jakob disease found no evidence of the disease (Evatt et al. 1998). ‘Look-back’ studies have not identified any cases of Creutzfeldt–Jakob disease developing in recipients who received blood from a donor in whom the disease was later diagnosed. The risk of CJD transmission through transfusion is now considered negligible, and plasma pools are no longer discarded if a contributing donor develops CJD. Deferrals designed to eliminate ‘high-risk’ donor categories, such as growth hormone and dura mater recipients, remain in force.
Variant Creutzfeldt–Jakob Disease
A neurodegenerative disease affecting cattle was first identified in England in 1984 and subsequently branded ‘mad cow disease’. Bovine spongiform encephalopathy (BSE) is now recognized as a prion-related disorder with more than 179 000 confirmed clinical cases of BSE, driven by the recycling of infection through the inclusion of bovine protein in cattle feed. A ban on feeding tissues from one ruminant animal to other ruminant animals, introduced in 1988, brought the British BSE epidemic under control. However, because of the long delay between infection and the onset of clinical signs of disease (5 years on average), the annual incidence of clinical cases did not peak until 1992. BSE has been found in several other European countries and in Canada. More rapid and accurate description of the epidemiology of BSE has been hampered by the absence of a diagnostic test that can be applied to live animals to detect those that are incubating the infection.
The human disease equivalent, new variant Creutzfeldt–Jakob disease (vCJD), was not identified until 1996, 7 years after the ban was introduced. A disease-related isoform of prion resembling that of BSE is consistent with the conclusion that BSE has crossed the species barrier, probably from ingestion of infected beef (Collinge and Rossor 1996; Bruce et al. 1997). Unlike classic CJD, vCJD usually affects younger subjects (age 18–53 years) and is characterized by early psychiatric and sensory symptoms. The disease progresses over 8 months to 3 years, with progressive dementia, ataxia and myoclonus. Brain biopsy has a characteristic pattern and Western blot analysis shows a diagnostic migration pattern of proteinase-treated PrPSc. Pathological PrPSc can be found in tonsils, spleen lymph nodes and appendix (Collinge et al. 1996). Prion protein gene analysis has shown that all cases were homozygous for methionine at codon 129 (Ironside and Head 2004). By October 2010, 222 definite and probable variant CJD cases had been reported worldwide in residents of 12 countries. The total numbers of clinical cases of variant CJD that will ultimately emerge remains uncertain, as does the frequency of long-term asymptomatic carriage during which apparently healthy individuals could transmit infection for example through blood donation or invasive medical procedures (deMarco et al. 2010). One model indicates that the primary epidemic in the susceptible genotype (methionine-methionine (MM)-homozygous at codon 129 of the prion protein gene) has now peaked, with an estimate of 40 future deaths (Ghani et al. 2003). However, the prevalence of vCJD infection in the UK population, estimated to be as high as 1 per 4000 based on a study of routinely acquired tonsils and appendices, raises the possibility of a second and third ‘wave’ of clinical cases in subjects heterozygous (methionine-valine) and homozygous for valine at codon 129.
Prion protein has been identified on a wide range of circulating cells including platelets, myeloid cells, lymphocytes and red cells (Dodelet and Cashman 1998; Holada and Vostal 2000; Bessos et al. 2001; Li et al. 2001). Findings in experimental models show that blood not only contains infective agents of prion diseases, but that no barrier to transmission exists with intraspecies transmission, and that the intravenous route of exposure to prions is fairly efficient (Casaccia et al. 1989; Cervenakova et al. 2003). The seminal experiments in sheep have shown transmission of BSE and scrapie by blood transfusion, and blood for transfusion in these experiments was obtained from sheep midway through the incubation period (Houston et al. 2000; Hunter et al. 2002). Infectivity has also been noted in the incubation period and symptomatic phase in a rodent model of vCJD (Cervenakova et al. 2003).
In 1997, the UK set up a surveillance system between the national CJD surveillance unit and the UK national blood services. In 2004, the first probable transfusion-associated case of vCJD was described (Llewelyn et al. 2004):
Related posts:
 Red cell antibodies against self-antigens, bound antigens and induced antigens
Red cell antibodies against self-antigens, bound antigens and induced antigens
 Blood grouping techniques
Blood grouping techniques
 Alternatives to allogeneic transfusion
Alternatives to allogeneic transfusion
 The transfusion of platelets, leucocytes, haematopoietic progenitor cells and plasma components
The transfusion of platelets, leucocytes, haematopoietic progenitor cells and plasma components
 The Rh blood group system (including LW and RHAG)
The Rh blood group system (including LW and RHAG)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


