- 2.1 Introduction
- 2.2 Normal resistance to infection
- 2.2.1 Non-specific resistance
- 2.2.2 Specific resistance
- 2.2.1 Non-specific resistance
- 2.3 Viral infection
- 2.3.1 Epstein–Barr virus infection
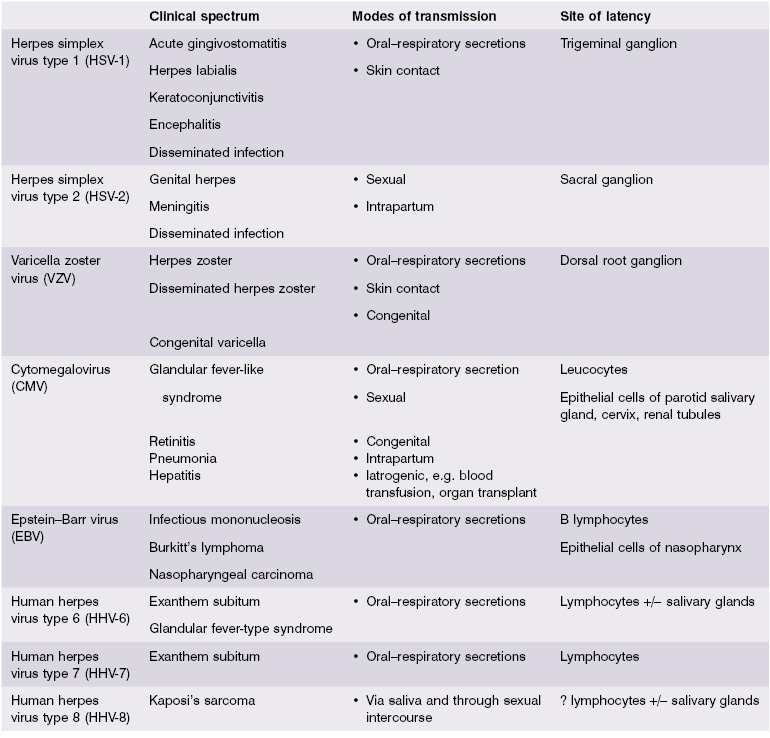
- 2.3.2 Herpes viruses in general
- 2.3.3 Direct effects of viruses
- 2.3.4 Viral strategies to evade the immune response
- 2.3.5 Bystander damage caused by the immune response to viral infection
- 2.3.6 Speculative effects of viral infection
- 2.3.1 Epstein–Barr virus infection
- 2.4 Bacterial infection
- 2.4.1 Normal immune responses to bacterial infections
- 2.4.2 Bacteria as superantigens
- 2.4.3 Bacterial evasion of immune defences
- 2.4.4 Bystander damage caused by the immune response to bacterial infection
- 2.4.1 Normal immune responses to bacterial infections
- 2.5 Mycobacterial infection
- 2.5.1 Mycobacterial infections
- 2.5.2 Mycobacteria and the normal immune response
- 2.5.3 Mycobacterial evasion of the immune response
- 2.5.4 Damage caused by the immune response to mycobacteria
- 2.5.5 Prevention of tuberculosis
- 2.5.1 Mycobacterial infections
- 2.6 Fungal infection
- 2.6.1 Mechanisms of immunity to fungal infections
- 2.6.2 Bystander damage caused by immune reactions to fungi
- 2.6.1 Mechanisms of immunity to fungal infections
- 2.7 Parasitic infection
- 2.7.1 Protozoal infection
- 2.7.2 Normal immune responses to protozoa
- 2.7.3 Protozoal evasion of immune responses
- 2.7.4 Helminth infections
- 2.7.5 Normal immunity to helminth infection
- 2.7.6 Helminth evasion of immune responses
- 2.7.7 Bystander damage caused by immune reactions to protozoa and helminths
- 2.7.1 Protozoal infection
 Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
2.1 Introduction
Infectious disease is the major cause of morbidity and mortality worldwide. In Africa alone, the World Health Organization estimates that about 100 million people suffer from malaria. New infectious diseases also attract attention in developed countries for several reasons. These include the identification of seemingly ‘new’ infections, such as Helicobacter pylori (recognized in 1989), new variant Creutzfeldt–Jakob disease (the causative prion was discovered around the same time), the epidemic of severe acute respiratory syndrome (SARS) (that spread from Hong Kong to infect individuals in 37 countries in early 2003) and the H1N1 and H5N1 (bird flu) influenza pandemics. There are also changes in clinical practice, which have altered patterns of hospital infection, such as antibiotic resistance and the increased spread of Clostridium difficile in hospitals. The growth in numbers of iatrogenically immunosuppressed patients receiving immunosuppression who are at risk from ‘opportunistic’ infections, as well as an increase in imported diseases accompanying the rising volume of international air travel, provide new challenges. There is also an increasing awareness of diseases resulting from self-damaging host responses to pathogens.
For most infections, a balance is maintained between human defences, including the immune system, and the capacity of the microorganism to overcome or bypass them (Table 2.1). A detailed discussion of virulence is outside the scope of this book, but disease will also occur if the host makes an inadequate or inappropriate immune response to an infection.
Table 2.1 Factors influencing the extent and severity of an infection
Pathogen factors
|
Host factors
|
2.2 Normal resistance to infection
2.2.1 Non-specific resistance
Non-specific or natural resistance refers to barriers, secretions and the normal flora that make up the external defences (Fig. 2.1), together with the actions of phagocytes and complement.
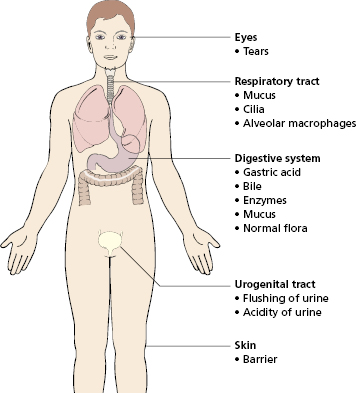
Mechanical barriers (Fig. 2.1) are highly effective, and their failure often results in infection; for example, defects in the mucociliary lining of the respiratory tract (as in cystic fibrosis) are associated with an increased susceptibility to lung infection. However, many common respiratory pathogens have evolved specific substances on their surfaces (e.g. the haemagglutinin of influenza virus), which help them attach to epithelial cells and so breach physical barriers.
Phagocytic cells ingest invading microorganisms and, in most cases, kill and digest them. There are two types of phagocyte: monocytes/macrophages and neutrophilic polymorphonuclear leucocytes (neutrophils). A prompt response to infection is achieved by having a population of cells that can be rapidly mobilized during an inflammatory response and being able to concentrate them at likely sites of infection.
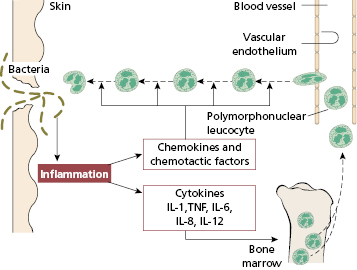
Neutrophils form a large circulating pool of phagocytic cells with reserves in the bone marrow. Invading microorganisms trigger an inflammatory response with the release of cytokines and chemotactic factors: as a result, circulating neutrophils adhere to vascular endothelium, squeeze out of blood vessels and actively migrate towards the focus of infection (Figs 1.13 and 2.2). Phagocytosis then occurs. Severe neutropenia or neutrophil dysfunction is associated with life-threatening infections, usually caused by common organisms such as Staphylococcus aureus, Gram-negative bacteria or fungi (see Chapter 3). Even in normal conditions, neutrophils are short-lived but produced in huge numbers: basal rate of production by the bone marrow of 5 × 1010–10 × 1010 neutrophils/day. In infection, the increased output from the bone marrow results in a neutrophilia, i.e. an excess of neutrophils in the blood. If a particularly rapid response is needed, immature cells may also be released – this was described as ‘a shift to the left’ on a blood film before CRP was available.
Fig. 2.2 A schematic representation of the mobilization of bone marrow stores of polymorphs following an inflammatory response.
Phagocytosis is promoted by serum factors termed ‘opsonins’ (see Chapter 1; Fig. 1.19): IgG antibody, complement and mannan-binding lectin are the best opsonins. Non-opsonized bacteria can still be recognized and bound by phagocyte receptors – pattern recognition receptors (PRRs) – particularly those that are specific for sugars present in bacterial cell walls (mannose receptor) but also to a variety of other pathogen-associated molecular patterns (PAMPS) (Fig. 2.3). Other receptors include CD14 that acts as a receptor for bacterial lipopolysaccharide (LPS); the integrin molecules CD11b/CD18, CD11c/CD18 that recognize several microbes including Leishmania, Bordetella, Candida and LPS and Toll-like receptors (TLRs) that recognize a range of pathogens (see section 1.2.2). Monocytes, dendritic cells and resident macrophages then initiate inflammation and also the adaptive immune response (see Chapter 1). Phagocytic receptors (FcIgRs) and complement receptors (C3R) are important for removal of bacteria before antigen-specific immune responses (T cells and antibody) have had a chance to develop.
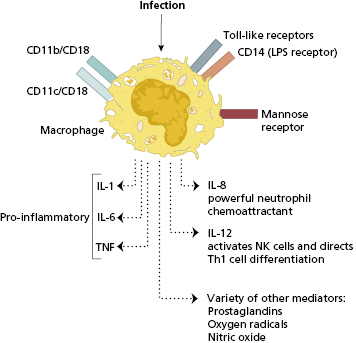
Resident macrophages occur in the subepithelial tissues of the skin and intestine and line the alveoli of the lungs. Organisms that penetrate an epithelial surface will encounter these local tissue macrophages (sometimes referred to as ‘histiocytes’). If invasion by microorganisms occurs via blood or lymph, then defence is provided by fixed macrophages lining the blood sinusoids of the liver (Kupffer cells), the spleen and the sinuses of lymph nodes. The interaction of macrophages with certain bacterial components leads to the production of an array of macrophage-derived cytokines, which non-specifically amplify inflammatory reactions (Fig. 2.3). Macrophages are able to engulf opsonized organisms as well as directly bind to certain pathogens by pattern recognition and other receptors.
Most pathogenic microorganisms have evolved methods of resisting phagocytic cells. Staphylococci produce potent extracellular toxins that kill phagocytes and lead to the formation of pus, so characteristic of these infections. Some microorganisms have substances on their cell surfaces that inhibit direct phagocytosis. Under these circumstances, phagocytosis can proceed effectively only when the bacteria are coated (opsonized) by IgG or IgM antibodies or complement. Other microorganisms, e.g. Mycobacterium tuberculosis, are effectively ingested by phagocytic cells but can resist intracellular killing.
2.2.2 Specific resistance
An antigen-specific immune response is conventionally classified into humoral and cell-mediated immunity (see Chapter 1). The relative importance of humoral vs. cell-mediated immunity varies from infection to infection. Experimental animal models and naturally occurring immunodeficiencies in humans (see Chapter 3) demonstrate that certain components of the immune response are essential for controlling particular infections (Fig. 2.4 and Fig 3.2).
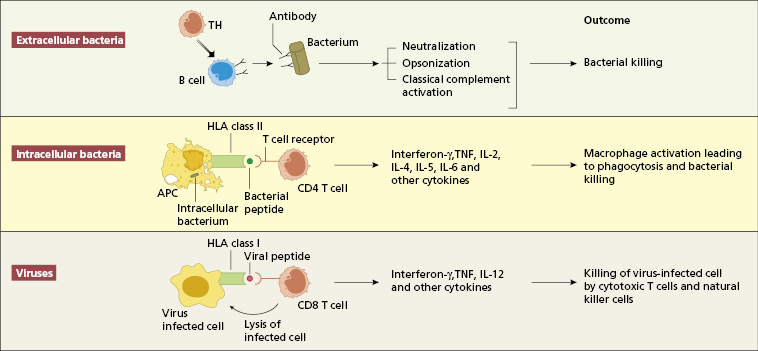
Individuals with antibody deficiency are prone to repeated infections with pyogenic bacteria (see Chapter 3), but immunoglobulin replacement therapy markedly reduces the frequency of these bacterial infections (Section 3.2.5). The course of infections with many viruses (such as varicella or measles) is normal in these patients.
T-lymphocyte function is more important than humoral immunity in both viral and intracellular bacterial infections. Patients with impaired cell-mediated immunity have difficulty in controlling and eradicating infections with viruses such as measles, varicella and herpes. They also show increased susceptibility to mycobacteria, pneumocystis, Listeria monocytogenes and some fungi. Recurrent viral or fungal infections or infection by an unusual organism suggest the possibility of an underlying T cell defect, either iatrogenically (as in chemotherapy or immunosuppression) or a (much rarer) primary immunodeficiency.
Infections are discussed by type of organisms in this chapter. It is impossible to discuss the immune response to all pathogens, so only illustrative examples are given in each section.
2.3 Viral infection
2.3.1 Epstein–Barr virus infection
Infectious mononucleosis is caused by the Epstein–Barr virus (EBV), a member of the herpes group of viruses. By the age of 3 years, 99% of children in developing countries have been infected subclinically with EBV. In developed countries, clinically recognizable infection most frequently occurs in the 15–25-year age group; the virus is excreted in oropharyngeal secretions for some months, and is responsible for person-to-person transmission.
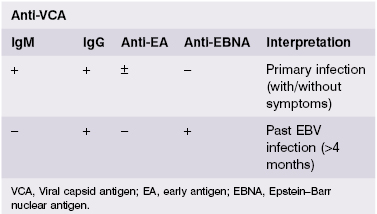
The pattern of antibody responses to different EBV antigens helps to distinguish acute or subclinical infection from past EBV infection (Table 2.2). IgM antibodies to the viral capsid antigen (VCA) appear early in the course of infection; by the time symptoms of infectious mononucleosis develop, IgG antibody titres to VCA are also high; however, testing of paired sera for a rise in antibody titre, used in the past for diagnosis of many viral infections, is not helpful. Antibodies to EB nuclear antigen (EBNA) develop about 4 months after infection and remain for life. Antibodies to early antigen (EA) appear during primary infection in about 70% of patients and WERE considered an indicator of active infection until superseded by real-time PCR (RT-PCR) and measurement of EBV-viral load for the early diagnosis of infectious mononucleosis, particularly in cases with inconclusive serological results.
EBV is unique among human viruses in that it produces disease by infecting and transforming B lymphocytes via the CD21 molecule on the B-cell surface. Infected B cells proliferate like tumour cells, and small numbers may produce free virus which can then transform other B lymphocytes. Up to half of the lymphoid cells from the tonsils of EBV-infected patients may be transformed. Primary EBV infection is stopped by two defences: a T-cell immune response capable of eliminating almost all virus-infected cells, and virus-neutralizing antibodies which prevent the spread of infection from one target cell to another. The characteristic ‘atypical lymphocytes’ are predominantly CD8+ cytotoxic T lymphocytes, which recognize and destroy EBV-infected B cells (Case 2.1).
 Case 2.1 Infectious mononucleosis
Case 2.1 Infectious mononucleosisA 20-year-old carpet fitter presented with a 1-week history of a sore throat, stiffness and tenderness of his neck, and extreme malaise. On examination, he was mildly pyrexial with posterior cervical lymphadenopathy, palatal petechiae and pharyngeal inflammation without an exudate. Abdominal examination showed mild splenomegaly. There was no evidence of a skin rash or jaundice.
The clinical diagnosis of infectious mononucleosis (‘glandular fever’) was confirmed on investigation. His white cell count was 13 × 109/l (NR 4–10 × 109/l) with over 50% of the lymphocytes showing atypical morphology (‘atypical lymphocytosis’). His serum contained IgM antibodies to Epstein–Barr viral capsid antigen (VCA), a common test for acute infectious mononucleosis (see Table 2.2). Liver function tests were normal.
He was treated symptomatically and was advised to avoid sporting activity until his splenomegaly had completely resolved, because of the danger of splenic rupture. Many patients show clinical or biochemical evidence of liver involvement and are recommended to abstain from alcohol for at least 6 months.
The importance of the immune response to EBV is illustrated by (i) rare patients with EBV-specific failure of immunity; and (ii) the occurrence of EBV-induced malignant transformation of B cells in patients receiving immunosuppressive therapy (see Case 7.2). In the first example, the X-linked ‘lymphoproliferative syndrome’ (XLP) affects males (aged 6 months to >20 years) who are unable to control EBV infection due to mutation in the gene encoding SAP (SLAM-associated protein). SAP mutations lead to a failure in signal transduction from the ‘signalling lymphocyte activation molecule’ (SLAM), which is present on the surface of T and B cells. Many patients with this syndrome die young unless they receive a human stem cell transplant; some die of lymphoma, some of aplastic anaemia, and others of haemophagocytic syndrome as part of their immunodeficiency.
Patients who receive immunosuppressive regimens, such as ciclosporin, antithymocyte globulin or monoclonal anti-T-cell antibodies following transplantation, also develop EBV tumours. These therapeutic agents are associated with EBV reactivation: about 1–10% of certain transplants are complicated by EBV-induced lymphoproliferative disease. Similarly, up to 2% of patients infected with human immunodeficiency virus (HIV) (Chapter 3) develop non-Hodgkin’s lymphoma but the incidence may be higher as many tumours are not found till post-mortem (see Case 3.10); EBV has been identified in most acquired immune deficiency syndrome (AIDS)-associated lymphomas.
Burkitt’s lymphoma is a highly malignant, extranodal tumour of B lymphocytes also strongly associated with EBV infection. It is endemic in certain African countries, where it represents approximately 90% of childhood cancers, in contrast to 3% in developed countries. The link between EBV and Burkitt’s lymphoma was substantiated by the demonstration of the EBV genome and EBV antigens in tumour cells. It is likely that Burkitt’s lymphoma is due to EBV-induced lymphoproliferation in individuals rendered susceptible by chronic malaria Proposed mechanisms for this failure to limit EBV infection include the ability of the malaria parasites to activate latently infected B cells with the emergence of a proliferating clone and exhaustion of EBV-specific T-cell responses after repeated malaria infections in children. EBV infection in this setting leads to chromosomal translocation(s) with consequent activation of the c-myc oncogene, resulting in lymphoma.
2.3.2 Herpes viruses in general
The herpes virus group consists of at least 60 viruses, eight of which commonly infect humans (Table 2.3). Two features of pathogenesis are common to all human herpes viruses. First, close physical contact must occur between infected and uninfected human individuals for transmission of virus and no other species is involved. ‘Close contact’ between cells, as occurs in blood transfusion or organ transplantation, provides potential routes of transmission for several herpes viruses – most notably cytomegalovirus (CMV). Second, after a primary infection, herpes viruses persist in the host throughout life.
To limit virus dissemination and prevent reinfection, the immune response must be able to stop virions entering cells, and to eliminate cells already infected in order to reduce virus shedding. Immunological reactions are thus of two kinds: those directed against the virion and those that act upon the virus-infected cell. In general, immune responses to the virion are predominantly humoral, while T-cell-mediated responses act on virus-infected cells. Antibodies and B cells see different epitopes from those seen by T cells. The major humoral mechanism involved is viral neutralization by antibody, but complement-mediated lysis of virus also occurs.
Viral neutralization prevents attachment of virus to target cell and is the function of IgG antibodies in the extracellular fluid, IgM in the blood and secretory IgA antibodies on mucosal surfaces. Only antibodies to those viral components responsible for attachment are neutralizing. The generation of antibodies of the correct specificity is therefore essential for an effective viral vaccine; antibodies to inappropriate antigens not only may fail to protect, but may actually provoke immune complex disease (see section 2.3.5).
Cell-mediated immunity is concerned with virus-infected cells rather than free virus. Virus-immune T lymphocytes recognize viral antigens in association with self-MHC class I glycoproteins (see Chapter 1). Cytotoxic T (Tc) cells lyse virally modified cells and limit disease by stopping production of infectious progeny, along with natural killer (NK) cells. T and NK cells are therefore concerned with recovery from virus infections; containment of the initial infection is mediated by type-I interferons (IFN-α and INF-β)(Fig. 2.5).
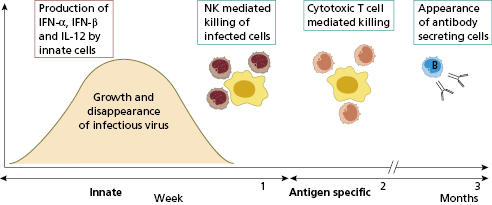
Most viral infections are self-limiting. Recovery from acute viral infections usually produces specific long-term immunity and secondary attacks by the same virus are uncommon and indicate immunodeficiency.
2.3.3 Direct effects of viruses
The clinical importance of viral infection depends not only on the number of host cells destroyed but also on the function of those cells. Destruction of relatively few cells with highly specialized function, such as neurotransmission or immunoregulation, can be disabling or life threatening. In contrast, destruction of larger numbers of less specialized cells, such as epithelial cells, has less drastic results. In order to gain entry into specialized cells, viruses interact with specific receptors on host cells – viral tropism. For example, EBV uses the C3d receptor (CD21), HIV uses multiple receptors (CD4, chemokine receptors: CCR5, CXCR-4) and the SARS-coronavirus (SARS-CoV) uses angiotensin-converting enzyme (ACE)-2 in order to gain entry into target cells.
Once inside a cell, a virus can kill the cell in several ways. Some viruses, such as poliovirus or adenovirus or their products, can block enzymes needed for cell replication or metabolism, while others may disrupt intracellular structures, such as lysosomes, releasing lethal enzymes. Some viral proteins inserted into the cell membrane can alter its integrity: measles virus, for instance, possesses fusion activity and causes cells to form syncytia. Some viruses can alter the specialized function of a cell without killing it. Usually, such cells belong to the central nervous or endocrine systems; dementia caused by HIV infection is an example of both atrophy and loss of function.
Transformation of host cells may occur with certain viruses that are potentially oncogenic (Table 2.4). Mostly, these are viruses that establish latency. Cells from Burkitt’s lymphoma, for instance, show a characteristic translocation between the long arms of chromosomes 8 and 14, suggesting that the tumour results from the translocation of the oncogene, c-myc, to an active region of the cellular genome (in this example, the locus for the immunoglobulin heavy chain). Human papilloma virus (HPV) oncogenicity is another example of viral transformation of host cells. The interaction of HPV E6 and E7 with critical cell cycle components is fairly well understood and leads to hyperplastic lesions (warts) or, when coupled with other mutations (e.g. through DNA damage through ultra-violet radiation), results in skin or cervical malignancies (Table 2.4).
Table 2.4 Viruses and malignant disease
| Malignancy | Virus |
|---|---|
| Certain T-cell leukaemias | Human T-cell leukaemia virus (HTLV-I) |
| Carcinoma of cervix | Herpes simplex (type 2) |
| Human papilloma viruses (HPV) | |
| Burkitt’s lymphoma | Epstein–Barr virus |
| Nasopharyngeal carcinoma | Epstein–Barr virus |
| Skin cancer | Human papilloma viruses (HPV) |
| Hepatocellular carcinoma | Hepatitis B |
| Hepatitis C | |
| Kaposi’s sarcoma | Human herpes virus 8 (HHV-8) |
Some viruses can interfere with the immune response by suppressing it or infecting cells of the immune system (Table 2.5). The best example of this phenomenon is AIDS (see Chapter 3), caused by HIV types 1 and 2 (HIV-1, HIV-2), which selectively infect and deplete CD4+ T lymphocytes and macrophages. The resulting profound immunosuppression leads to the development of the severe, disseminated, opportunistic infections and tumours that characterize AIDS. Measles virus is a less well known but important example. Before widespread measles immunization and treatment for tuberculosis (TB) were available, outbreaks of acute measles infection were associated with the reactivation and dissemination of miliary TB, due to reduced T-cell-mediated immunity.
Table 2.5 Examples of viruses infecting cells of the immune system
| Cell | Virus | Outcome |
|---|---|---|
| B lymphocytes | Epstein–Barr virus | Transformation and polyclonal B-cell activation |
| T lymphocytes | Measles | Replication in activated T cells |
| Human T-cell leukaemia virus I | T-cell lymphoma/leukaemia | |
| Human immunodeficiency virus 1 and 2 | Acquired immune deficiency syndrome | |
| Macrophages | Dengue | |
| Lassa | Viral haemorrhagic fevers | |
| Marburg – Ebola |
2.3.4 Viral strategies to evade the immune response
Viruses have evolved ingenious mechanisms for evading or interfering with immune responses. An important viral strategy for evasion is entry into a latent state. All human herpes viruses can remain latent (see Table 2.3), undergoing periodic cycles of activation and replication. The viral genome remains within the host cell but no expression of viral antigens occurs. When the equilibrium between virus and host defence is upset, perhaps by other infections, metabolic disturbances, ageing or immunosuppression, the virus is no longer controlled, with subsequent expression of disease.
Varicella zoster (VZV) is a good example of a disorder caused by a virus that normally lies latent in the dorsal root ganglion but becomes active when host immunity is impaired, to cause shingles as in Case 2.2, following prolonged corticosteroid therapy. Other herpes viruses may remain latent in other anatomically defined sites (see Table 2.3), for instance, herpes simplex virus in the trigeminal ganglion (producing ‘cold sores’) and activated by sunlight, stress or inter-current infection.
 Case 2.2 Recurrent herpes zoster
Case 2.2 Recurrent herpes zosterA 72-year-old woman was commenced on oral corticosteroids for giant cell arteritis. Over the next 6 months she had three episodes of a painful, vesicular rash (‘shingles’) typical of herpes zoster affecting the ophthalmic division of the right trigeminal nerve. Each episode was successfully treated with oral acyclovir, but she experienced considerable post-herpetic neuralgia. A steady improvement in her arteritic symptoms and inflammatory indices allowed a reduction in steroid dosage over a period of 6 months, with no further episodes of zoster.
Other mechanisms of evading immune responses are given in Table 2.6. Antigenic variation is best illustrated by influenza A, an RNA virus surrounded by a lipid envelope with two inserted proteins – haemagglutinin and neuraminidase – against which most neutralizing antibodies are directed. The virus can evade antibody responses by modifying the structure of these proteins in two ways: antigenic drift and antigenic shift. Antigenic drift is minor structural change caused by point mutations altering an antigenic site on haemagglutinin. Such mutations probably account for the ‘minor’ epidemics of influenza occurring most winters. Antigenic shift is a major change in the whole structure of haemagglutinin or neuraminidase, which has caused periodic influenza pandemics in the past.
Table 2.6 Mechanisms of immune evasion by viruses
| Mechanism | Example |
|---|---|
| Non-expression of viral genome | Herpes simplex virus (latent in neurones) |
| Production of antigenic variants | Influenza, HIV |
| Inhibition of MHC expression | Adenovirus Cytomegalovirus |
| Mimicking MHC | Cytomegalovirus |
| Suppress NK activity | Cytomegalovirus |
| Induction of lymphocyte apoptosis | Cytomegalovirus |
| Production of inhibitory cytokines | EBV and IL-10 |
HIV, Human immunodeficiency virus; EBV, Epstein–Barr virus; IL-10, interleukin-10.
Viral persistence
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


