INTRODUCTION
The indolent non-Hodgkin lymphomas (NHLs) represent approximately one-third of all malignant lymphomas (1,2); most are of B-cell lineage. Follicular lymphoma (FL) is the most common indolent lymphoma. Other indolent B-cell lymphomas include small lymphocytic lymphoma/chronic lymphocytic leukemia (SLL/CLL), the marginal zone B-cell lymphomas (MZLs; extranodal, nodal, and splenic), and lymphoplasmacytic lymphoma (LPL), most cases of which are more specifically classified as Waldenström macroglobulinemia (WM) (Table 7-1) (2). Mantle cell lymphoma can morphologically resemble indolent B-cell lymphomas, but it is often clinically more aggressive and therefore is not covered in this chapter. Indolent T-cell NHLs, such as mycosis fungoides, are also not covered in this chapter.
| Entities included | Follicular lymphoma Small lymphocytic lymphoma/chronic lymphocytic leukemia Extranodal marginal zone B-cell lymphoma (MALT lymphoma) Splenic B-cell marginal zone lymphoma Nodal marginal zone lymphoma Lymphoplasmacytic lymphoma (including Waldenström macroglobulinemia) |
| Age | Mostly a disease of older adults (usually over the age of 40 years) |
| Extent of disease | Often disseminated (except MALT lymphoma), with >80% having stage III-IV disease. Bone marrow involvement common |
| Natural history | Low proliferation fraction. Slow-growing; may have a waxing and waning course. Patients typically survive for many years. Transformation to large cell lymphoma can occur. |
| Curability | Although current therapy such as radiotherapy or chemotherapy can often control the disease, it usually fails to eradicate the tumor except for early-stage disease (including MALT lymphoma). This is reflected in a continuous downward slope of relapse-free survival curves for patients with these lymphomas. |
FOLLICULAR LYMPHOMAS
Follicular lymphomas, the second most commonly occurring lymphoma in the United States, represents 22% of all NHLs (1) and 80% of indolent B-cell lymphomas. Follicular lymphoma occurs almost exclusively in adults, with an equal frequency in men and women. The incidence rates are highest among Caucasians, and median age at diagnosis is approximately 58 years (3). Risk of FL has been shown to be increased in persons with a first-degree relative with NHL or who worked as spray painters and among women with Sjögren syndrome (3). Of FL cases, 2% to 3% transform annually to diffuse large B-cell lymphoma (DLBCL) (4). Survival for patients with FL is improving, with a median survival of 8 to 10 years in the prerituximab era (2,5,6); in more modern eras, median survival has been reported to be greater than 18 years (7).
Patients with FL most often present with asymptomatic lymphadenopathy. Constitutional symptoms such as fever, drenching night sweats, and significant weight loss occur in approximately 15% of patients. Patients may have symptoms related to lymph node enlargement, especially when there are bulky masses in the retroperitoneum. Other symptoms can include fatigue and, occasionally, end-organ consequences such as obstructive uropathy or bone marrow compromise. Central nervous system (CNS) disease is rare. Urgent situations, such as superior vena cava syndrome or spinal cord compression, are rare, in part related to the usual slow pace of growth of lymphadenopathy in FL. Spontaneous regression of lymphadenopathy can occur in FL. Such regressions, however, are usually partial and are typically short-lived. The potential of FL to wax and wane provides one of several clues that suggest that the host immune system can play an important role in the disease course in FL. Consequently, FL has been a prime focus for immunotherapy approaches.
Approximately 80% to 90% of patients with FL present with advanced-stage disease (stage III or IV) with generalized lymphadenopathy. The bone marrow is involved in approximately 50% of patients. Clinical features suspicious for transformation to DLBCL include rapidly progressive lymphadenopathy, systemic (B) symptoms, localized pain, and a rise in serum lactate dehydrogenase (LDH) level.
In FL, the normal lymph node architecture is partially or completely replaced by lymphoma, which typically forms follicles but rarely can be diffuse, composed of centrocytes (small, cleaved cells) and centroblasts (large, noncleaved cells). The method currently recommended in the World Health Organization (WHO) classification to grade FL is based on a count of the centroblasts (2,8). In grade 1 FL, centroblasts are rare, less than 5 per ×400 microscopic field. Grade 2 FL contains 5 or more but less than 15 centroblasts per ×400 microscopic field. The current WHO classification states that there is no prognostic benefit derived from distinguishing grade 1 from grade 2 cases and designates these tumors as FL grade 1 or 2. At least two types of grade 3 FL are described. In grade 3a, more than 15 centroblasts per ×400 microscopic field are present. In grade 3b, sheets of centroblasts are present with rare or absent centrocytes (2). Recent data suggested that FL grade 3b has more features in common with DLBCL than with the indolent FL (9).
Some patients with B-cell lymphoma of germinal center cell origin may have histologic discordance (ie, low-grade FL in the bone marrow and DLBCL in a lymph node) (10). In addition, different lymph nodes biopsied in a given patient or a single lymph node can comprise different grades of FL.
Follicular lymphoma is a neoplasm of mature B-cell lineage. Most grade 1 and 2 tumors express immunoglobulin (Ig), but a subset of FLs, mostly grade 3, may be Ig negative. All FLs express pan B-cell markers and typically express Ig and B-cell antigens at high density (“bright” immunofluorescence by flow cytometry). These neoplasms also express the germinal center-associated markers CD10, Bcl-6, and LMO2 and are negative for T-cell antigens. Bcl-2 is expressed in 80% to 90% of FLs but can be negative, most often in grade 3 neoplasms (2). Because Bcl-2 is negative in reactive germinal centers, this marker is helpful in differential diagnosis (Fig. 7-1).
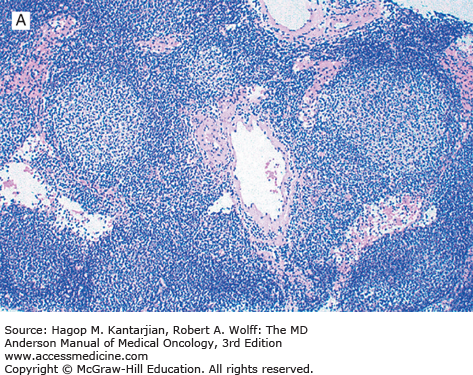
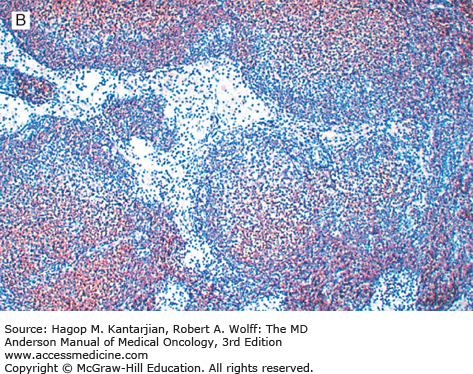
Using conventional cytogenetic analysis, approximately 75% of FL cases grow in culture and can be successfully karyotyped. The cytogenetic hallmark of FL is t(14;18)(q32;q21), which is identified in 80% to 90% of cases. A small subset of FLs lack the t(14;18), suggesting that a minor pathway of follicular lymphomagenesis may exist that is independent of t(14;18). This appears to apply particularly to grade 3b nodal FL, FLs arising in extranodal sites such as skin, and rare FLs that occur in children, which commonly lack the t(14;18).
As a result of the t(14;18), the Bcl-2 oncogene on chromosome 18q21 is translocated adjacent to the joining region of the immunoglobulin heavy chain (IgH) gene on chromosome 14q32. The Bcl-2 gene is deregulated by being placed under the influence of IgH gene regulatory elements (enhancer region). Insights about the role of the Bcl-2 gene in FL were a gateway to the identification of a large family of proapoptotic and antiapoptotic genes, which play a role in a wide variety of hematopoietic and solid neoplasms (11,12,13).
The breakpoints on chromosome 18 are primarily clustered at two sites, the major (MBR) and minor (mcr) breakpoint cluster regions, involved in 50% to 60% and 10% to 20% of cases, respectively (14). Other breakpoint clusters have also been described, for example, the intermediate cluster region (ICR). The ICR is involved in approximately 5% of cases; there may be geographic variations in the frequencies of t(14;18) breakpoints (15).
The Bcl-2 protein is a 25-kDa molecule that is overexpressed in FL and protects cells from programmed cell death (apoptosis) (11,12,16). Inhibition of apoptosis prolongs cell life, resulting in an expanded compartment of B cells, thereby providing more opportunity for additional molecular defects, which presumably are involved in histologic transformation. The presence of the t(14;18) alone appears not to be sufficient for neoplastic transformation. The t(14;18) has been identified in rare cells in the tonsils and lymph nodes of normal individuals without clinical evidence of lymphoma (17).
Other cytogenetic abnormalities have been reported in FL. Of these, trisomy 7 and 18, abnormalities of 3q27-28 and 6q23-26, and 17p deletions are most frequent. Abnormalities of 3q27-28 involve the Bcl-6 gene and most often occur in the form of translocations. Secondary cytogenetic and molecular genetic abnormalities have also been extensively studied, including in the context of transformation of FL to DLBCL (18,19,20).
The importance of the immunologic microenvironment in the clinical behavior of FL has been an area of intensive study. Gene expression profiling methods have shown molecular signatures attributable to subsets of T cells and macrophages in FLs that influence the risk of disease progression and prognosis (21,22,23,24).
Follicular lymphoma in situ (FLIS) is thought to be a preneoplastic or possibly very early stage of FL (25). Most often, FLIS is an incidental finding that occurs in lymph nodes excised for a variety of reasons (eg, axillary lymph nodes in a patient with breast carcinoma). The overall frequency of FLIS is low, approximately 2%.
Morphologically, FLIS is difficult to recognize in routine, tissue sections stained with hematoxylin-eosin. The lymph node architecture is normal, with widely scattered follicles that are of normal size and inconspicuous. The germinal centers have a sharp peripheral margin and are monotonous, composed almost exclusively of centrocytes, a morphologic clue to the diagnosis of FLIS (26). Immunohistochemical analysis is essential for the recognition of FLIS. The germinal center cells are strongly positive for BCL2 and CD10 (26,27). Typically, BCL2 expression by the FLIS cells is brighter than the expression level by mantle zone cells surrounding the germinal center. The cells of FLIS also express pan B-cell antigens and other germinal center B-cell markers, such as BCL-6 and LMO2. Cytogenetic and molecular studies have shown that the cells of FLIS carry t(14;18)(q32;q21) and monoclonal Ig gene rearrangements. Array comparative genomic hybridization and other methods performed on FLIS lesions have shown that the cells of FLIS carry the t(14;18) but have relatively few secondary genetic abnormalities, in contrast with fully developed FL (28). EZH2 mutations have been detected in FLIS, suggesting this is another early lesion in FL pathogenesis.
Evidence of overt lymphoma at another anatomic site also is present in a small subset of patients with FLIS (26). In addition, some patients with FLIS subsequently develop a histologically discordant type of lymphoma. These cases suggest that FLIS could be a marker of genetic instability or a marker of genetic predisposition to lymphoma. Lymphomas most often reported in association with FLIS include classical Hodgkin lymphoma, splenic marginal zone lymphoma, and SLL/CLL (27).
In aggregate, the data suggest that FLIS is a neoplastic process representing an early step in FL pathogenesis that is unlikely to affect patient survival if it is an isolated finding. However, because a subset of some patients with FLIS also has or may develop overt lymphoma at other anatomic sites, staging studies are indicated. If FLIS is the only disease discovered, overtreatment is to be avoided.
Clinical evaluation requires a comprehensive history, including age; sex; presence of B symptoms (fevers, chills, drenching night sweats, unexplained weight loss of more than 10% of body mass over 6 months, pruritus); fatigue; and a history of malignancy. Physical examination should include identification and measurement of all accessible lymph node sites, including epitrochlear and occipital lymph nodes, and assessment of the abdomen for splenomegaly or hepatomegaly.
The diagnosis is best established by an excisional lymph node biopsy to provide adequate tissue for assessment of the lymph node architecture. The most easily accessible lymph node may not be the most informative or representative one. For example, if a small peripheral lymph node shows grade 1 FL but the patient has a large abdominal mass, a high serum LDH level, and other features suggestive of transformation, then an additional biopsy to exclude higher-grade disease should be considered because this would influence the selection of appropriate therapy. Core needle biopsies guided by radiographic or imaging techniques may be performed on masses that are not easily accessible. Fine-needle aspiration (FNA) can be misleading for initial diagnosis; complete classification may not be possible because the limited tissue sample prevents the assessment of architecture, and there is the possibility of sampling error. In the initial staging evaluation, FNA may play a role in documenting and defining sites of involvement.
Once the diagnosis of FL has been established, patients should undergo testing to determine stage, assess prognostic risk factors, and evaluate their general health. A complete blood cell count may show anemia or thrombocytopenia, which can result from bone marrow involvement or occasionally from hypersplenism or autoimmune problems. Leukemic involvement can occur in 10% of patients. Serum LDH and β2-microglobulin (B2M) levels may be elevated and are of prognostic significance. Bilateral bone marrow biopsies with unilateral aspiration are recommended for the staging workup because of the patchy nature of involvement. In FL, the bone marrow characteristically shows a paratrabecular pattern of involvement (Fig. 7-2). Because the lymphoma cells are associated with stroma and are not easily aspirated, bone marrow aspirate smears assessed by routine light microscopy may not be informative. Flow cytometry and molecular assessment (eg, polymerase chain reaction, PCR) of aspirate material can increase the sensitivity of bone marrow assessment, but in the absence of morphologic abnormalities, positive PCR or flow findings are traditionally not taken as evidence to warrant assignment of stage IV (29). For example, it is well established that patients at Ann Arbor stage I or II can have cells with the t(14;18) detected in the peripheral blood or bone marrow by PCR.
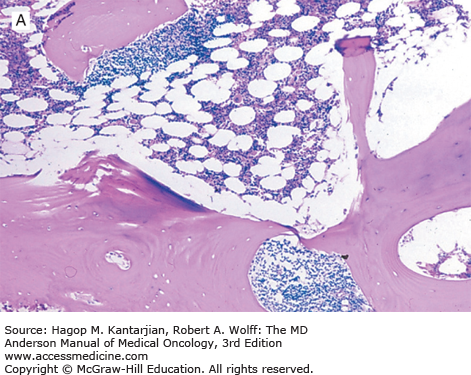
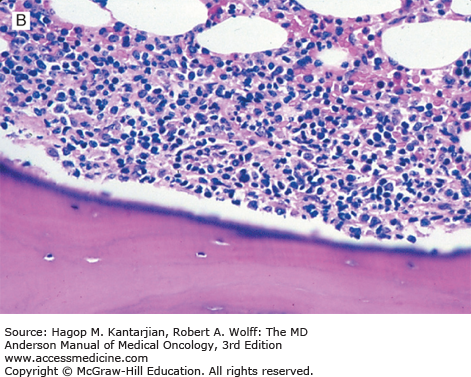
Imaging studies should include neck and chest computed tomography (CT) for delineation of lymphadenopathy. Abdominal and pelvic CT scans are essential for assessing lymphadenopathy as well as organomegaly. Positron emission tomography (PET) using fluorine-18 fluorodeoxyglucose (FDG) is a useful tool for assessing patients with FDG-avid lymphomas and Hodgkin lymphoma. The field is evolving, and PET combined with CT (PET-CT) is replacing PET alone. Compared with CT scans, PET-CT can improve the accuracy of staging for nodal and extranodal sites and leads to a change in stage in 10% to 30% of patients, more often upstaging these patients (30). Improving staging accuracy is important to ensure patients are appropriately treated. Although most lymphomas are FDG avid, there is great variability in FDG uptake (31,32,33). Imaging by PET can be less reliable in indolent lymphoma. Therefore, adoption of PET scanning for staging patients with indolent lymphoma has not been embraced to the same extent that it has for DLBCL and Hodgkin lymphoma. Recent consensus guidelines recommend the use of PET-CT for staging in clinical practice and clinical trials for patients with FL and may be used to select the best site to biopsy.
The prognostic importance of histologically distinguishing grade 1 or 2 FL from the more aggressive grade 3 FL is well accepted. However, most investigators have not found a clear difference in long-term survival between patients with grades 1 and 2 FL, although older literature suggested that FL grade 2 (nodular mixed-cell lymphoma, in older nomenclature) was more prone to early progression than grade 1 if therapy was deferred (34). Higher degrees of nodularity have been associated in some reports with improved outcome. An increased proliferation rate is associated with a poorer prognosis (35,36).
Variables that have been shown to correlate with survival in patients with FL include tumor burden, host factors, and response to therapy. Tumor burden can be estimated by assessing stage of disease, size of nodal disease, bone marrow involvement, serum B2M, LDH levels, and number of nodal sites. Adverse host factors include advanced age, B symptoms, low hemoglobin level, male gender, and poor performance status. The background cells in the diagnostic lymph node biopsy may also provide prognostic information, as shown in gene expression profiling studies (22,24).
The International Prognostic Index (IPI) was devised for aggressive lymphomas and consists of five variables: age, performance status, Ann Arbor stage, extranodal involvement, and serum LDH level (37). The IPI is also a useful predictor of survival in patients with indolent B-cell lymphomas. One important limitation of this system is that only 11% of the patients fell into the high-risk group, and most of these patients had poor performance status and would be poor candidates for aggressive therapy.
Partly for that reason, a Follicular Lymphoma International Prognostic Index (FLIPI) was developed. Initially, an eight-parameter model (age ≥60 years, male gender, Ann Arbor stages III and IV, nodal sites ≥5, bone marrow involvement, serum LDH level greater than normal, hemoglobin level <12 g/dL, and lymphocytes ≥1000/mL) was proposed (38). However, a simplified version of this model was found to be comparably predictive, using the five parameters of age, Ann Arbor stage, serum LDH, hemoglobin level, and number of nodal sites (39). This prognostic model separates patients into three prognostic groups: good risk with 0 to 1 factors, intermediate risk with 2 factors, and poor risk with 3 or more factors. The overall 5-year survival was 90% for the good-risk group, 78% for the intermediate-risk group, and 53% for the poor-risk group (39). The validity of the FLIPI model has been demonstrated in rituximab-treated patients (40). A FLIPI-2 model was developed through the prospective collection of prognostic data, producing a five-factor model that incorporates age (>60 years adverse); hemoglobin level (<12 g/dL adverse); serum B2M level (adverse if above normal range); bone marrow (adverse if involved); and size of lymphadenopathy (>6 cm adverse), which has not been validated (41).
There are more similarities than differences in these models. For instance, the importance of serum B2M was shown in the univariate analyses of both the IPI and FLIPI data sets, but the B2M data were collected prospectively only in the FLIPI-2 report. The importance of sampling the bone marrow, as shown in the FLIPI-2 report, deserves particular emphasis, because practice patterns indicate that the important data offered are often not collected. Easily applied models such as the IPI, FLIPI, or FLIPI-2 can provide a framework for selecting the timing or intensity of therapy, and can facilitate the interpretation of clinical trials, by providing a tool to assess for disparities in patient selection when results of various trials are compared (Table 7-2).
At the time of relapse, favorable predictors for survival in patients with FL include having achieved a complete response with initial therapy, having had a durable remission of more than 1 year following initial therapy, and being less than 60 years of age.
An international working group has recommended end-of-treatment assessment and defined response criteria for NHL. These criteria have recently been updated, with important modifications, including incorporation of PET-CT (30). In FDG-avid FL, PET-CT should be used for response assessment, using the 5-point scale (1, no uptake above background; 2, uptake ≤ mediastinum; 3, uptake > mediastinum but ≤ liver; 4, uptake moderately > liver; 5, uptake markedly higher than liver and/or new lesions). The criteria based on PET-CT eliminate complete response unconfirmed (CRu) evaluation and improve the prognostic value of a partial response (PR). A complete metabolic response (PET-CT), even with a persistent mass, is considered a complete remission (CR). With CT-based assessment, a complete radiologic response is determined by nodal masses regressing to 1.5 cm or less in the longest diameter and no extralymphatic sites of disease. Bone marrow reevaluation is performed to confirm clinical remission if the bone marrow was initially positive.
Although the monitoring of patients with molecular studies is not currently considered standard practice, detection of the t(14;18)/IGH-BCL2 by PCR techniques has been useful in the monitoring of subclinical disease. “Molecular remission,” the disappearance of cells with the t(14;18) detected by PCR, used to be considered a rarity in patients with FL treated with standard therapy. Gribben et al reported that only 1 of 212 (0.5%) patients achieved molecular remission following conventional chemotherapy (42). With high-dose therapy and stem cell transplantation (SCT), however, molecular remissions could be attained, and those with molecular remission experienced more durable remissions. Improvements in therapy have changed this picture. Even before the advent of anti-CD20 monoclonal antibody (MAb) therapy, more potent chemotherapy regimens were capable of inducing molecular remission in over half of patients. With the availability of rituximab, which can largely eradicate B cells from the blood and bone marrow, it is now common to see molecular remission. Some recent studies continued to show that molecular remission correlates with more durable clinical remission(43,44), but other studies did not (45).
There are limited data supporting patient benefit from surveillance imaging in FL. As a consequence, there is considerable variation in practice. Zinzani et al performed one of the only prospective studies of surveillance imaging, which included 78 patients with FLs in first CR following induction therapy (46). Patients received PET-CT scans every 6 months for 2 years, with annual scans thereafter. The relapse rate was 8% to 10% as shown by scans performed until 36 months and decreased after that time. As the purpose of the study was to describe specificity of scans, the impact on management and subsequent outcome was not reported. Gerlinger et al performed surveillance with annual CT scans and bone marrow biopsies in 71 patients with FL in second or subsequent remission following SCT (47). Although approximately half of relapses were CT detected, few such patients required immediate treatment; there was no difference in overall survival between patients in whom disease relapse was detected by imaging compared with clinical presentation. Their conclusion was surveillance imaging was futile in this setting. Surveillance CT scans result in radiation exposure, patient anxiety, and significant health-care costs, which should be carefully weighed against any potential benefit. The National Comprehensive Cancer Network (NCCN) guidelines suggest “surveillance CT scan up to every 6 months for the first 2 years following completion of therapy, and not more than annually thereafter.” In contrast, the European Society for Medical Oncology (ESMO) guidelines are more conservative, suggesting minimal adequate radiological or ultrasound investigations every 6 months for 2 years and annually thereafter. Regular CT scans are not mandatory outside clinical trials, and PET-CT should not be used for surveillance (48).
At diagnosis, approximately 15% to 20% of patients with FL have limited-stage disease (stages I and II). This stage of disease is associated with a favorable outcome, and up to half of these patients may be cured. Consequently, seizing the opportunity for cure should be strongly considered, even though some advocate deferral of therapy (49). Several series have reported long-term disease-free survival of approximately 35% to 50% for stage I to II patients treated with involved-field radiotherapy (RT), so it appears that many of these patients are cured (49,50,51,52,53). Studies with extended-field or total lymphoid RT, in an attempt to increase cure rates, have not shown convincing additional benefit.
The integration of chemotherapy with involved-field RT has shown promising results in some trials. Investigators at MD Anderson Cancer Center (MDACC) prospectively treated 85 patients with stage I or II FL with 10 cycles of COP-Bleo (cyclophosphamide, vincristine, prednisone, and bleomycin) or CHOP-Bleo (COP-Bleo plus doxorubicin) and involved-field RT “sandwiched” after the third cycle. The disease-free survival at 5 and 10 years was 80% and 72%, respectively—an apparent improvement over results with RT alone (54). Analysis of the National LymphoCare Study also demonstrated improvement in progression free survival (PFS) in patients who received systemic therapy in combination with RT (55). The role of rituximab in stage I or II disease is not clearly defined; efficacy has been extrapolated from the advanced-stage literature. A prospective, randomized study is being conducted here at MDACC to investigate the addition of rituximab to RT in limited stage, previously untreated patients with FL; results are yet to come.
In summary, patients with limited-stage FL appear to be potentially curable. The role of RT in these patients is well established, so involved-field RT remains the standard treatment (see Table 7-2). Despite the established role of involved-field RT and its endorsement by experts, RT appears to be underutilized in practice (55,56). Total lymphoid RT and combined-modality therapy approaches remain controversial and are seldom used.
For decades, the treatment of patients with advanced stage FL has been built on two pillars that lean in opposite directions. First, there are numerous effective therapeutic options that can induce remission (Table 7-3); second, relapse appears to be inevitable. If and when therapeutic advances lead to more comprehensive control of FL, then there may be consensus about early intervention because a smaller tumor burden would presumably be more easily treatable, analogous to the stage I or II situation. Until then, it is still the case that deferral of therapy is a common consideration for many asymptomatic patients with low tumor burden.
| Stages I-II | IF RT RT and CTa |
| Stages III-IV | Deferral of therapy (if no threatening disease) Single-agent alkylators Single-agent MoAb COP and variants, with MoAb CHOP and variants,a with MoAb FND and variants,a with MoAb RT (stage III) CT and RTa (stage III) Consolidation (see text) Maintenance (see text) |
When treatment is deemed appropriate, there are a number of considerations in choosing an initial management strategy. With advances in supportive care, we have seen improvement in the tolerability of chemotherapy and SCT. With a better understanding of B-cell lymphomagenesis and the role of the microenvironment, noncytotoxic therapeutic strategies are being incorporated into front-line clinical trials. The range of therapeutic options for patients with advanced-stage FL remains broad (see Table 7-3). The therapeutic goals continue to focus on minimizing toxicity, providing palliation, and attaining durable CR. Consideration of a clinical trial when available is advisable.
For patients with asymptomatic FL with a low tumor burden, deferment of therapy until the development of symptoms or high tumor burden has been accepted based on three randomized trials demonstrating survival equivalent to that with immediate therapy in the prerituximab era (57,58,59). With the long-term safety and efficacy of rituximab established, observation has been challenged in the modern era with rituximab monotherapy in patients with low tumor burden. The goal is to delay the first cytotoxic chemotherapy and to have an impact on health-related quality of life.
The findings of a randomized trial of rituximab versus a watch-and-wait approach in asymptomatic, nonbulky patients with FL suggested patients who received rituximab monotherapy had a delay in progression in comparison to those with an initial watch-and-wait approach (60). Those who received rituximab maintenance following rituximab induction also reported improved quality of life. No significant difference in overall survival was reported.
The RESORT study was a randomized cooperative group trial evaluating two dosing strategies of rituximab monotherapy in patients with untreated, grade I or II FL of low tumor burden (61). There were 289 patients randomized to either rituximab maintenance or retreatment after responding to rituximab induction (4 weekly doses). The median time to treatment failure (FFS) was similar (3.9 vs 4.3 years), quality of life was similar, and significant toxicity was infrequent among both strategies. For patients with FL with a low tumor burden, rituximab monotherapy followed by a retreatment strategy was associated with less rituximab use while providing comparable disease control to that achieved with a maintenance strategy.
For patients with untreated FL of low tumor burden not eligible for a clinical trial or deemed appropriate for more intensive therapy, an initial management strategy of rituximab monotherapy was associated with a delay in the need for cytotoxic therapy, was well tolerated, and was perceived by some to enhance their health-related well-being. Hence, the decision between observation and rituximab monotherapy should be made following a detailed discussion about the goals of treatment.
Several trials have shown convincingly that the addition of rituximab to chemotherapy leads to improved outcomes (Table 7-4). Incorporation of rituximab (R) has become standard. Suitable partners for rituximab in induction regimens include cyclophosphamide, vincristine, and prednisone (CVP); cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP); bendamustine (B); and fludarabine-based regimens (such as FND [fludarabine, mitoxantrone, and dexamethasone]) (57,60,61,62,63,64,65,66
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


