Therapy for chronic lymphocytic leukemia (CLL) has evolved dramatically throughout the years. In 1997, rituximab (Rituxan), a CD20 monoclonal antibody (mAb), became the first mAb approved by the Food and Drug Administration for marketing in the treatment of cancer, specifically targeting B-cell malignancies. Over the last 10 years, rituximab or other mAbs including alemtuzumab and ofatumumab have become an integral part of the standard of care for CLL patients as single agents or in combination with chemotherapy or other immunotherapy. This review discusses the currently approved and novel mAbs for the treatment of CLL.
Key points
- •
Standard chemotherapeutic options for CLL cause significant immune suppression and myelosuppression, are not well-tolerated by the elderly population and have not consistently offered survival advantage and therefore, alternative immune therapies have been developed, including various monoclonal antibodies (mAbs).
- •
MAbs mechanisms of cytotoxicity include complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity, antibody-dependent cell phagocytosis, and direct apoptosis.
- •
Rituximab was the first mAb approved by the FDA for marketing in the treatment of cancer, and over the last 10 years, rituximab and other mAbs including alemtuzumab and ofatumumab have become integral parts of the standard of care for CLL patients as single agents or in combination with chemotherapy or other immunotherapy.
- •
Following the introduction of rituximab, multiple subsequent novel mAbs have been designed to target CLL, are at various stages of development, and will continue to revolutionize therapy for CLL patients.
Introduction
Chronic lymphocytic leukemia (CLL) is the most common adult leukemia in the United States and Europe. CLL is predominantly a cancer of the elderly, with a median age at diagnosis of CLL of 72 years, and approximately 70% of patients are newly diagnosed over the age of 65 years. Despite recent advances in the field, CLL is a cancer that is considered incurable outside the setting of a stem cell transplant. The other approved, standard regimens available to treat symptomatic CLL patients are used with a goal of prolonging the time between treatment (treatment-free survival [TFS]). In recent years, many studies have identified select genomic and cellular features that help predict response to therapy and TFS, such as presence of del(17p13.1) and del(11q22.3) on interphase cytogenetics, complex karyotype by metaphase cytogenetics, immunoglobulin variable heavy chain (IgVH) mutational status, elevated β2-microglobulin, and presence of ZAP70 expression or methylation. Furthermore, at present there is no proven survival benefit with early therapy for CLL and, therefore, patients are observed until they become symptomatic from their leukemia.
Therapy for CLL has significantly changed over the years, from an emphasis on alkylator-based (chlorambucil and cyclophosphamide) therapies in the 1970s to fludarabine-based therapies in the early 1990s. These therapies can be toxic, with accompanying immunosuppression and myelosuppression in the majority of CLL patients who are generally elderly with multiple medical morbidities. In addition, these advances in treatment have modest effectiveness in patients with high-risk genomic features, such as del(17p13.1), p53 mutations, ZAP-70-unmethylated disease, and IgVH-unmutated disease These factors prompted continued exploration of new therapeutic strategies for CLL.
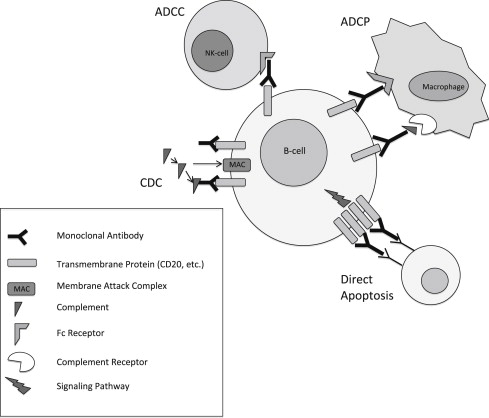
For more than 3 decades the application of monoclonal antibodies (mAbs) as targeted therapy against cancer has been explored (as reviewed by Scott and colleagues ). mAbs have a fixed effector cell-binding region (Fc) and a variable region with affinity toward a specific antigen that can provide considerable selectivity. Tumor elimination can be mediated by mAbs through various mechanisms, including antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), complement-dependent cytotoxicity (CDC), and direct cell toxicity through transmembrane signaling. ADCC is an immune reaction whereby the targeted tumor cell is coated with antibodies and subsequently destroyed by effector immune cells (ie, natural killer cells or macrophages) via cell lysis following cell membrane puncture by perforin and release of granzyme B or other cytotoxic cytokines. ADCP occurs when antibody-coated cells are engulfed by macrophages and related cells. CDC is an immune reaction whereby the formation of antibody-antigen complexes initiates the complement cascade wherein effector immune cells disrupt the target tumor cell surface membrane, causing water and solutes to exit the cell and thus mediating lysis of the tumor cell. These different mechanisms of cell killing are illustrated in Fig. 1 .
In 1997, rituximab (Rituxan) became the first therapeutic antibody approved for marketing in CD20-positive low-grade B-cell non-Hodgkin lymphoma (NHL). Since then, significant advances have been made with both single-agent and combination therapies including rituximab in CLL and other B-cell malignancies. Because of the acclaimed success of this mAb, a significant amount of preclinical and clinical research has been produced with alternative CD20 mAbs and other target proteins found on the B-cell surface membrane ( Table 1 ). The focus of this article is to review the laboratory concepts and clinical data available regarding mAb therapy for CLL ( Table 2 ).
| Protein | Cell Types | Notably Not Present |
|---|---|---|
| CD20 | Pre-B to mature B cells | Plasma cells |
| CD52 | Most B and T cells, granulocytes, monocytes, macrophages, eosinophils, NK cells, and dendritic cells | Plasma cells |
| CD37 | Pre–B cells to mature B cells, Minimally T cell, NK cells, monocytes | Plasma cells Platelets Red blood cells |
| CD40 | B cells (including plasma), monocytes, dendritic cells, CD8 T cells, epithelial and endothelial cells, platelets | — |
| CD19 | B cells, follicular dendritic cells | Plasma cells |
| HLA-DR | B cells, monocytes/macrophages, dendritic cells, thymic epithelial cells, and activated T cells and endothelial cells | — |
| Antibody | Target | Antibody Characteristics | Comparison with Rituximab | Status |
|---|---|---|---|---|
| Rituximab | CD20 | Type I, mouse/human chimeric IgG1 | N/A | FDA approved |
| Ofatumumab | CD20 | Type I, human IgG1 | More effective CDC | FDA approved |
| Obinutuzumab (GA-101) | CD20 | Type II, humanized, IgG1 | More potent ADCC and DCC, no CDC | Phase I/II |
| Veltuzumab | CD20 | Type I, humanized IgG1 | Better off rate and CDC, enhanced binding avidity | Phase I |
| Tru-016 | CD37 | SMIP, mouse/human chimeric, IgG1 | Superior ADCC and DCC | Phase I |
| 37.1 | CD37 | SMIP, mouse/human chimeric, IgG1, engineered Fc portion | Superior DCD, clearance of CLL cells from whole blood samples | Preclinical |
| 37.2 | CD37 | SMIP, humanized, IgG1, engineered Fc portion | Superior DCD, clearance of CLL cells from whole blood samples | Preclinical |
| Dacetuzumab (SGN-40) | CD40 | Humanized, IgG1 | Unknown | Phase I |
| Lucatumumab (HCD-122) | CD40 | Humanized, IgG1 | Superior ADCC | Phase I |
| XmAb5574 | CD19 | Human, IgG1, engineered Fc portion | Superior ADCC | Phase I |
| MDX-1342 | CD19 | Human, nonfucosylated Fc portion | Unknown | Phase I |
| Apolizumab (Hu1D10) | HLA-DR | Humanized murine, IgG1 | Unknown | Discontinued after phase I |
| IMMU-114 | HLA-DR | Humanized murine, IgG4 | Superior DCD | Preclinical |
Introduction
Chronic lymphocytic leukemia (CLL) is the most common adult leukemia in the United States and Europe. CLL is predominantly a cancer of the elderly, with a median age at diagnosis of CLL of 72 years, and approximately 70% of patients are newly diagnosed over the age of 65 years. Despite recent advances in the field, CLL is a cancer that is considered incurable outside the setting of a stem cell transplant. The other approved, standard regimens available to treat symptomatic CLL patients are used with a goal of prolonging the time between treatment (treatment-free survival [TFS]). In recent years, many studies have identified select genomic and cellular features that help predict response to therapy and TFS, such as presence of del(17p13.1) and del(11q22.3) on interphase cytogenetics, complex karyotype by metaphase cytogenetics, immunoglobulin variable heavy chain (IgVH) mutational status, elevated β2-microglobulin, and presence of ZAP70 expression or methylation. Furthermore, at present there is no proven survival benefit with early therapy for CLL and, therefore, patients are observed until they become symptomatic from their leukemia.
Therapy for CLL has significantly changed over the years, from an emphasis on alkylator-based (chlorambucil and cyclophosphamide) therapies in the 1970s to fludarabine-based therapies in the early 1990s. These therapies can be toxic, with accompanying immunosuppression and myelosuppression in the majority of CLL patients who are generally elderly with multiple medical morbidities. In addition, these advances in treatment have modest effectiveness in patients with high-risk genomic features, such as del(17p13.1), p53 mutations, ZAP-70-unmethylated disease, and IgVH-unmutated disease These factors prompted continued exploration of new therapeutic strategies for CLL.
For more than 3 decades the application of monoclonal antibodies (mAbs) as targeted therapy against cancer has been explored (as reviewed by Scott and colleagues ). mAbs have a fixed effector cell-binding region (Fc) and a variable region with affinity toward a specific antigen that can provide considerable selectivity. Tumor elimination can be mediated by mAbs through various mechanisms, including antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), complement-dependent cytotoxicity (CDC), and direct cell toxicity through transmembrane signaling. ADCC is an immune reaction whereby the targeted tumor cell is coated with antibodies and subsequently destroyed by effector immune cells (ie, natural killer cells or macrophages) via cell lysis following cell membrane puncture by perforin and release of granzyme B or other cytotoxic cytokines. ADCP occurs when antibody-coated cells are engulfed by macrophages and related cells. CDC is an immune reaction whereby the formation of antibody-antigen complexes initiates the complement cascade wherein effector immune cells disrupt the target tumor cell surface membrane, causing water and solutes to exit the cell and thus mediating lysis of the tumor cell. These different mechanisms of cell killing are illustrated in Fig. 1 .
In 1997, rituximab (Rituxan) became the first therapeutic antibody approved for marketing in CD20-positive low-grade B-cell non-Hodgkin lymphoma (NHL). Since then, significant advances have been made with both single-agent and combination therapies including rituximab in CLL and other B-cell malignancies. Because of the acclaimed success of this mAb, a significant amount of preclinical and clinical research has been produced with alternative CD20 mAbs and other target proteins found on the B-cell surface membrane ( Table 1 ). The focus of this article is to review the laboratory concepts and clinical data available regarding mAb therapy for CLL ( Table 2 ).
| Protein | Cell Types | Notably Not Present |
|---|---|---|
| CD20 | Pre-B to mature B cells | Plasma cells |
| CD52 | Most B and T cells, granulocytes, monocytes, macrophages, eosinophils, NK cells, and dendritic cells | Plasma cells |
| CD37 | Pre–B cells to mature B cells, Minimally T cell, NK cells, monocytes | Plasma cells Platelets Red blood cells |
| CD40 | B cells (including plasma), monocytes, dendritic cells, CD8 T cells, epithelial and endothelial cells, platelets | — |
| CD19 | B cells, follicular dendritic cells | Plasma cells |
| HLA-DR | B cells, monocytes/macrophages, dendritic cells, thymic epithelial cells, and activated T cells and endothelial cells | — |
| Antibody | Target | Antibody Characteristics | Comparison with Rituximab | Status |
|---|---|---|---|---|
| Rituximab | CD20 | Type I, mouse/human chimeric IgG1 | N/A | FDA approved |
| Ofatumumab | CD20 | Type I, human IgG1 | More effective CDC | FDA approved |
| Obinutuzumab (GA-101) | CD20 | Type II, humanized, IgG1 | More potent ADCC and DCC, no CDC | Phase I/II |
| Veltuzumab | CD20 | Type I, humanized IgG1 | Better off rate and CDC, enhanced binding avidity | Phase I |
| Tru-016 | CD37 | SMIP, mouse/human chimeric, IgG1 | Superior ADCC and DCC | Phase I |
| 37.1 | CD37 | SMIP, mouse/human chimeric, IgG1, engineered Fc portion | Superior DCD, clearance of CLL cells from whole blood samples | Preclinical |
| 37.2 | CD37 | SMIP, humanized, IgG1, engineered Fc portion | Superior DCD, clearance of CLL cells from whole blood samples | Preclinical |
| Dacetuzumab (SGN-40) | CD40 | Humanized, IgG1 | Unknown | Phase I |
| Lucatumumab (HCD-122) | CD40 | Humanized, IgG1 | Superior ADCC | Phase I |
| XmAb5574 | CD19 | Human, IgG1, engineered Fc portion | Superior ADCC | Phase I |
| MDX-1342 | CD19 | Human, nonfucosylated Fc portion | Unknown | Phase I |
| Apolizumab (Hu1D10) | HLA-DR | Humanized murine, IgG1 | Unknown | Discontinued after phase I |
| IMMU-114 | HLA-DR | Humanized murine, IgG4 | Superior DCD | Preclinical |
CD20 antibodies
CD20 is a protein expressed on the cell membrane of early pre–B cells and remains through B-cell development until postgerminal center cells differentiate into plasma cells. CD20 is present on both resting and activated, and malignant and nonmalignant B cells, making it an ideal target for B-cell malignancies. Of note, CLL cells express dim CD20 on their surface in comparison with normal B cells and most other B-cell malignancies. The function of CD20 is still unknown, although studies derived from genetically engineered mice demonstrate that B-cell development and function is normal in the absence of this protein.
Antibodies directed against CD20 have been classified into 2 major types, termed type I and type II. Type I antibodies show stronger C1q binding and potent induction of CDC, which may be related to inducing segregation of CD20 into plasma membrane lipid rafts. A lipid raft is a microdomain on the plasma membrane consisting of glycophospholipids and protein receptors that are more organized and tightly packed than the surrounding bilayer membrane, but float freely in the membrane bilayer. It is unclear as to why the translocation of CD20 into lipid rafts influences the antibody’s ability to activate complement in such a significant manner. One theory is that the concentration of the antibodies into a comparatively small area of the plasma membrane provides an ideal density for the juxtaposed Fc regions to engage the C1q heads, thereby triggering the classic complement cascade. Another theory is that the raft provides a cholesterol-rich environment that is advantageous for complement activation through C3 deposition or by providing a region of the membrane that is more susceptible to penetration by the membrane attack complex (MAC). In addition, type I antibodies cause low levels of direct signaling and cell death, with the requirement of a cross-linking antibody to mediate this process. By contrast, type II antibodies exhibit reduced binding to C1q, do not segregate CD20 into lipid rafts, and therefore cause lower levels of CDC. However, type II antibodies cause potent induction of direct cell death without requiring a cross-linking antibody. Rather, type II antibodies activate a caspase-independent, lysosomal-dependent mechanism of cell death that is dependent on homotypic adhesion. For a direct comparison of type I and type II antibodies, see Table 3 .
| Characteristic | Type I | Type II |
|---|---|---|
| Complement-dependent cytotoxicity | +++ | + |
| Antibody-dependent cell-mediated cytotoxicity and phagocytosis | +++ | +++ |
| Direct cell cytotoxicity | + | +++ |
| Localization of CD20 on lipid rafts | + | − |
The contribution of different mechanisms of antibody killing in CLL by rituximab has been extensively explored and is still viewed as controversial. The authors’ group was the first to demonstrate true in vivo caspase-dependent apoptosis and also downregulation of antiapoptotic proteins in CLL patients having a rapid blood response to rituximab. Beum and colleagues have demonstrated evidence of in vivo CDC activation and CD20 shaving (trogocytosis) in CLL, and have advocated for reduced dosing of antibody to avoid depleting complement. Although Fc receptor engagement by effector cells (ADCC and ADCP) has been shown to be important in many in vivo animal models, surrogate evidence of the importance of these mechanisms is lacking in CLL, based on there being no correlation of response with FcγR-IIa and FcγR-IIIa polymorphisms associated with high-affinity binding and enhanced effector cell activation. In addition, CLL has intrinsic immune defects as a consequence of both immunosuppressive cytokines and ligands that diminish natural killer (NK)-cell, monocyte, and macrophage dysfunction.
Rituximab
Rituximab, the first approved antibody for cancer treatment, is a chimeric murine/human type I monoclonal immunoglobulin G1 (IgG1) directed against CD20. Rituximab mediates CDC, ADCC, and direct cell apoptosis in the treatment of B-cell malignancies.
Rituximab monotherapy
The initial landmark phase III trial, which earned approval for single-agent use of rituximab (375 mg/m 2 intravenously [IV] for 4 weekly doses) in NHL, included 33 patients with small lymphocytic lymphoma (SLL). Only 12% of these SLL patients achieved response (all partial response [PR]). Several smaller studies (reviewed by Jaglowski and Byrd ) showed similar modest clinical responses with single-agent rituximab in CLL patients, and it was unclear at that point whether rituximab would continue to be developed for CLL/SLL patients. Lower responses in CLL as opposed to NHL were thought to be secondary to diminished CD20 expression, altered innate immune function, or different pharmacokinetic features of CLL in comparison with NHL. However, subsequent trials using higher doses of rituximab administered either 3 times weekly or at higher weekly dosing (up to 2250 mg/m 2 per dose) showed improved responses in relapsed CLL patients with a response duration similar to those seen in NHL. These trials paved the way for further study of rituximab in CLL. Subsequently, a phase II trial using rituximab (375 mg/m 2 ) weekly for 4 doses in the front-line treatment of CLL showed a better response in the previously untreated patients (overall response rate [ORR] 51%) compared with the refractory/relapsed patients in previous trials (ORR 13%). These trials prompted combinations of rituximab with chemotherapy (chemoimmunotherapy) in an attempt to further improve efficacy in CLL patients.
Rituximab chemoimmunotherapy
Multiple phase II studies have combined rituximab with various chemotherapeutic agents to show improved response. This section reviews the high-impact trials. Rituximab was combined with fludarabine (FR) in refractory (n = 11) and treatment-naïve (n = 20) CLL patients in a phase II trial that demonstrated an ORR of 87% with 32% complete response (CR). Another phase II trial (CALGB-9712) evaluated the difference between the concurrent or sequential administration of fludarabine and rituximab in 104 treatment-naïve patients with CLL. The concurrent cohort experienced more severe hematologic and infusion-related toxicities, but demonstrated an ORR of 90% (CR 47%) compared with an ORR of 78% (CR 28%) in the sequential cohort. Subsequent review of CALGB-9712 indicated that those patients with normal cytogenetics had a better 3-year overall survival (OS) (86%) than those patients with del(17p13.1) (33%) or del(11q22.3) (53%). The retrospective comparison between patients on this trial and a similarly designed study (CALGB-9011), which used fludarabine alone, demonstrated that the patients who received FR had a significantly improved progression-free survival (PFS) and OS compared with those who received fludarabine alone. Long-term follow-up of the patients on CALGB-9712 did not show an increased risk of therapy-related myeloid neoplasms. Another phase II study using sequential fludarabine and rituximab in 60 treatment-naïve CLL patients demonstrated an ORR of 93% (CR 78%), providing further evidence of this regimen’s activity in CLL.
Rituximab in combination with fludarabine and cyclophosphamide (FCR) has been thoroughly investigated. In a large study, 300 treatment-naïve CLL patients who received FCR demonstrated an impressive ORR of 95% (CR 72%) and a 6-year PFS and OS of 51% and 77%, respectively. Most common adverse events were cytopenias and infections. There were 8 patients who developed therapy-related myelodysplasia. In another large study, 177 relapsed/refractory CLL patients demonstrated an ORR of 73% (CR 25%). Median time to progression was 28 months. Common grade 3/4 toxicities were neutropenia (81%) and infection (16%). Patients with del(11q22.3) appeared to benefit from the addition of cyclophosphamide to fludarabine and rituximab as opposed to continued inferior outcomes in patients with del (17p13.1) and IgVH-unmutated CLL.
Pentostatin, a nucleoside analogue that is less myelotoxic than fludarabine, has been studied in combination with rituximab and cyclophosphamide (PCR). A study of PCR in 32 relapsed/refractory CLL patients demonstrated an ORR of 75% (CR 25%). Common toxicities included myelosuppression and infection A front-line study of PCR in 65 CLL patients demonstrated an ORR of 91% (CR 41%). The patients with del(11q22.3) again demonstrated that the addition of cyclophosphamide negates the poor prognosis associated with the marker. The most common adverse events were again myelosuppression and infection.
The alkylator-like molecule bendamustine has recently been successfully combined with rituximab (BR) in CLL. In a newly published phase II study, 117 treatment-naïve CLL patients received BR and demonstrated an ORR of 88% (CR 23%). At a median observation time of 27 months, event-free survival was 34 months and 90.5% of the patients were still living at the time of publication. Patients with del(11q22.3), trisomy 12, del(17p13.1), and IgVH-unmutated disease demonstrated response to BR at rates of 90%, 95%, 38%, and 89%, respectively. Grade 3/4 adverse events observed were thrombocytopenia (22.2%), neutropenia (19.7%), anemia (19.7%), and infection (7.7%). The same group reported the preliminary results of a phase II study of 81 relapsed/refractory patients who received BR and achieved an ORR of 77% (CR 15%; 62 patients were evaluable). Severe toxicities included myelosuppression and infection, with 3 patients dying of infections related to neutropenia caused by treatment. Longer-term analysis of this study is pending. A randomized phase III study comparing BR with FCR in front-line CLL therapy is ongoing (CLL10 Trial).
For rituximab chemoimmunotherapy in phase III studies, 2 landmark trials confirmed improved response rates and OS with FCR in both front-line and relapsed/refractory CLL patients. In the CLL8 trial, 817 CLL patients were randomized to receive either FCR or FC. The ORR was significantly better in the FCR group, at 90% versus 80% in the FC group ( P <.0001). The median PFS was significantly improved in the FCR group, at 51.8 months versus 32.8 months in the FC group ( P <.0001; hazard ratio [HR] 0.56; 95% confidence interval [CI] 0.46–0.69). In addition, the 3-year estimated OS was significantly improved in the FCR group, at 87% in the FCR group versus 83% in the FC group ( P = .012; HR 0.76; 95% CI 0.48–0.92). No increase in the number of infections was noted with the addition of rituximab, and more patients on the FC arm died during the trial. In the relapsed/refractory setting, the REACH trial randomized 552 CLL patients who had received a median of one line of therapy (mostly single-agent alkylator therapy) to receive either FCR or FC. The ORR was significantly improved in the FCR group, at 69.9% versus 58.0% in the FC group ( P = .0034). The median PFS was significantly improved in the FCR group, at 30.6 months versus 20.6 months in the FC group ( P <.001; HR 0.65; 95% CI 0.51–0.82). Hematologic toxicity was the most significant adverse event. These 2 studies have placed rituximab as a standard addition to front-line and subsequent lines of therapy for CLL.
Rituximab immunomodulation therapy
Lenalidomide is an immunomodulatory drug, which is active as a single agent in CLL. In vitro, the agent enhances NK-cell–mediated ADCC of rituximab against CLL cells, making lenalidomide an attractive agent to use in combination with rituximab in clinical trials (LR). In a phase II trial, 59 relapsed/refractory CLL patients received LR and demonstrated an ORR of 66% (CR 10%). Of note, the patients with del(17p13.1) (n = 15) in the study demonstrated an ORR of 53%. Reported 2-year OS was 83%. There were 3 patient deaths while on therapy that were thought to be unrelated to the study drug. Two second malignancies were observed (1 colon cancer and 1 myelodysplastic syndrome). The most common grade 3/4 adverse events were neutropenia (47%), infection (31%), thrombocytopenia (22%), anemia (10%), and 1 tumor lysis syndrome. Tumor flare reactions (27%) were all grade 2 or less. In the front-line setting, a phase-II trial, 2-arm study of 69 CLL patients (arm A = patients <65 years old, arm B = patients ≥65 years) who received LR has been reported. At the time of publication, 57 patients were available for analysis. Preliminary results of the study indicate an ORR of 94% (CR 20%) in arm A and 77% (CR 9%) in arm B. Neutropenia was the most common grade 3/4 adverse event. Infusion and tumor flare reactions were more frequent in the first cycle. A lower proportion of arm B patients were able to complete 7 full cycles of therapy (16 of 27) compared with those in arm A (35 of 39), mainly secondary to toxicity. Pending final publication of the study, this regimen appears to be efficacious and tolerable in CLL.
Rituximab maintenance therapy
Rituximab has been studied as a maintenance regimen for patients who achieved response with standard chemoimmunotherapy regimens. In one phase II study, 75 patients received 4 weekly doses of rituximab (375 mg/m 2 ) as maintenance therapy following response to fludarabine. Those patients who continued to have minimal residual disease (MRD) then received 4 monthly doses of rituximab at 375 mg/m 2 followed by 12 monthly doses at 150 mg/m 2 . The MRD patients receiving consolidation had a significantly longer 5-year PFS than the patients who did not receive consolidation (87% vs 32%, P = .001). Several ongoing phase III trials are assessing the efficacy of maintenance rituximab in CLL ( www.clinicaltrials.gov ), and this approach should still be viewed as investigational.
Rituximab therapy for the autoimmune complications of CLL
Another proposed use of rituximab in CLL is for the autoimmune complications that commonly occur with CLL, including autoimmune hemolytic anemia (AIHA), immune thrombocytopenia (ITP), and pure red cell aplasia. Rituximab was initially described in the treatment of steroid-refractory pure red cell aplasia and AIHA. Subsequently, successful treatment of 2 patients with pure red cell aplasia with rituximab (375 mg/m 2 ) weekly for 2 weeks was described. Eight patients with steroid-refractory AIHA were treated with rituximab and dexamethasone, and all patients achieved remission of AIHA. Retreatment was also successful for relapses in this group. In a case series of CLL patients with AIHA treated with rituximab, 12 of 14 patients had increased hemoglobin levels. In the ITP setting, 3 CLL patients who developed ITP while on fludarabine therapy and who were refractory to both steroids and intravenous immunoglobulin were treated with rituximab for 4 weekly doses. These patients all had a rapid and durable response to rituximab therapy. Although there are limited randomized data, these reports in conjunction with the experience at the authors’ institution provide evidence for rituximab’s effectiveness in the setting of autoimmune complications of CLL.
In general, rituximab is very well tolerated. The most common adverse event is infusion-related toxicity, and although more than 90% of patients experience some type of infusion-related reaction, it is generally mild to moderate in severity. Patients with CLL and those with an increased number of circulating lymphocytes are at a higher risk of the more severe adverse events, such as respiratory insufficiency and tumor lysis syndrome. Rare cases of hepatitis B or JC polyomavirus reactivation have been reported.
Ofatumumab (Arzerra, HuMax-CD20)
Because of the success of rituximab, many other CD20 antibodies have been developed to enhance select mechanisms of killing. Ofatumumab is the other currently approved type I humanized CD20 IgG1 antibody. In contrast to rituximab, it binds to a different epitope of CD20, which is composed of both the small and large extracellular loops of the CD20 molecule that facilitates enhanced CDC, direct killing (with cross-linking), and ADCC against CLL cells in comparison with rituximab. Fig. 2 depicts the binding sites of rituximab and ofatumumab on the CD20 protein. Ofatumumab has mediated CDC against rituximab-resistant Raji cell and CLL cells with low expression of CD20 in vitro. The agent appears to cause more potent CDC, has a slower off rate, and more stable CD20 binding than rituximab. The improved CDC may be the result of the greater proximity of the small-loop binding site to the cell membrane, which could potentially lead to more effective deposition of complement on the cell surface.
Related posts:
 The Significance of Stereotyped B-Cell Receptors in Chronic Lymphocytic Leukemia
Phosphoinositide 3′-Kinase Inhibition in Chronic Lymphocytic Leukemia
The Significance of Stereotyped B-Cell Receptors in Chronic Lymphocytic Leukemia
Phosphoinositide 3′-Kinase Inhibition in Chronic Lymphocytic Leukemia
 The Role of Minimal Residual Disease Measurements in the Therapy for CLL
Genomic Approaches to Chronic Lymphocytic Leukemia
Biology of Chronic Lymphocytic Leukemia in Different Microenvironments
Phosphoinositide 3′-Kinase Inhibition in Chronic Lymphocytic Leukemia
The Role of Minimal Residual Disease Measurements in the Therapy for CLL
Genomic Approaches to Chronic Lymphocytic Leukemia
Biology of Chronic Lymphocytic Leukemia in Different Microenvironments
Phosphoinositide 3′-Kinase Inhibition in Chronic Lymphocytic Leukemia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


