Mechanisms of glioma-media immunosuppression. Glioma cells produce immunosuppressive cytokines (interleukin-10 [IL-10] and tumor growth factor-beta [TGF-β]) that downregulate the antitumor activity of T cells and natural killer (NK) cells. Specifically, these cytokines inhibit an inflammatory phenotype and induce regulatory T-cell differentiation. Glioma cells are also known to directly evade immune recognition through decreased expression of surface major histocompatibility complex (MHC) molecules, and through direct apoptotic effects on cytotoxic T cells via PDL1/PD-1 interactions.
This treatment mode is further limited by the passivity of the instigated immunity, with duration of immune response tethered to the half-life of the delivered agent. Persistent treatment effect can develop, but we have learned more and more that this likely depends on the recruitment of subsequent T-cell immunity. Some contemporary antibody therapies then aim to solicit and direct T cells not otherwise specific for tumor by employing bispecificity for a tumor target and the T-cell receptor (bispecific T-cell engagers or BiTEs). These therapies in MG date back to 1990,22 with more contemporary versions rapidly progressing through preclinical testing.23 These also are discussed further in later sections.
More direct focus on T cells has been the teaching borne out of multiple immunotherapeutic modalities, as these are the effectors of the desired adaptive, cytotoxic immunity. More commonly, then, vaccine-based therapies for glioblastoma multiforme (GBM) have employed DC (reviewed by Fecci et al.),24,25 with the goal of simultaneously and potently teaching, directing, and activating nascent T-cell responses against tumor. Most have demonstrated some level of efficacy in phase I/II studies. Logistics involve DC production from autologous blood products, ex vivo loading with selected or unselected tumor antigens, activation/maturation, and reinjection with the aim of migration to lymph nodes and priming of the nascent T-cell response. Necessarily, such vaccine production is highly customized to patient, tedious, and non-standardized with regard to methods for each of the above logistical components. Furthermore, the reliance upon nascent T cells, which suffer rather excessive tumor-induced deficits, is limiting. Finally, there is an incredible complexity of DC subsets, and the appropriate subset(s) to isolate for autologous infusion as well as the optimal methods of activation remain areas of important study. DCs continue as one of the most tried methods of tumor vaccination, however, and form a presumed mainstay of sipuleucel-T, the lone FDA-approved antitumor (prostate) vaccination strategy to date.1Although the trials with sipuleucel-T proffered a number of lessons that can be extrapolated to brain tumor immunotherapy, its approval and proprietary production methods have done little to provide the answers to aforementioned questions regarding standardized strategies for DC-based immunization, perhaps preventing an organized advance of this approach within the clinical arena.
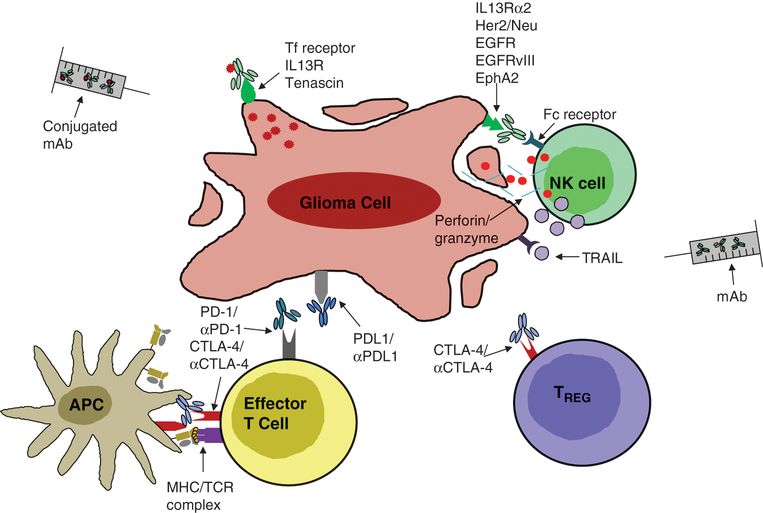
Antibody-based immunotherapies for malignant glioma. Antibodies can be engineered to bind tumor-associated antigens expressed on the glioma cell surface (e.g., EGFR, EGFRvIII, IL13R). Binding by these antibodies triggers antibody-dependent cell cytotoxicity (perforin/granzyme and cytotoxic cytokines) through Fc-receptor binding on innate immune cells. Such antibodies can also be conjugated to toxins, thereby directly and specifically targeting small-molecule therapy to tumor cells. Additionally, antibodies can be made specific for surface receptors on T cells through which signaling is known to have an immunosuppressive effect during antigen priming (e.g., CTLA-4) or effector function (e.g., PD-1), thereby abrogating the inhibitory effect triggered upon ligand binding.
Whereas DC and antigenic vaccines have the inevitable goal of T-cell stimulation, autologous T cells can also be directly harvested, trained/expanded/activated ex vivo against tumor, and transferred back to patients either alone or in conjunction with other so-handled immune cells, such as DC. In their earlier renditions, these ALTs included the transfer of a variety of immune populations, not just T cells. These have included peripheral blood mononuclear cells; lymphokine/mitogen-activated killer cells; tumor-infiltrating lymphocytes; and CTL, administered either systemically or into the tumor cavity (reviewed by Fecci and Sampson).19 Targets have varied, and newer renditions have combined ALT with active vaccination26 in efforts to promote survival and functional expansion of the transferred cells in vivo (NCT0114427, NCT01801852). Beyond ensuring cell survival, an additional “rate-limiting step” for ALT therapy has been the generation of large numbers of functional tumor-specific T cells ex vivo. Contemporary solutions to both challenges are discussed in the Emerging Topics section of this chapter.
Other lessons accumulated through immunotherapy’s adolescence relate to highlighted weaknesses in our capacity for judging our successes and failures. Classically, researchers have applied World Health Organization (WHO) criteria, and more recently, Response Evaluation Criteria In Solid Tumors (RECIST) criteria27 for evaluating responses to cancer therapies. Terms such as complete response, partial response, mixed response, stable disease, and progressive disease are strictly defined and used to evaluate therapies (typically chemotherapy). As noted above, a frequent pattern seen in immunotherapy trials has been an unaltered time to progression, yet improved overall survival (OS). This discordance accompanies the common findings of disease regression following an initial progression, or reductions in overall tumor burden despite the appearance of new lesions. These “immunotherapeutic patterns” are not frequently observed with cytotoxic therapies. WHO/RECIST criteria mandate that such patients be included in the progressive disease group when results are reported, necessarily hindering statistical analyses and diluting observed therapeutic effects. Furthermore, progressive disease might require ceasing an experimental therapy. As a result, groups such as the Cancer Immunotherapy Consortium of the Cancer Research Institute have since attempted to design more appropriate “immune-response criteria” to be applied during immunotherapy trials.28–30 These are based more on tumor burden and broaden the types of observed patterns (including stable disease) that might qualify as response to therapy, in an effort to permit reasonable alignment between clinical outcome (OS) and reported responses to therapy. These have not been universally accepted, but continue to expand in recognition and use.
Evidence-based practice
Cancer immunotherapy and, particularly, brain tumor immunotherapy remain young and growing fields. As such, there currently exist no level-one data supporting implementation. What does exist, however, is an expanding network of clinical trials aimed at supplying rationale, discovery, and optimization.
Rationale and discovery: clinical trials
Heat shock proteins: the alliance consortium
Heat shock proteins (HSP) are molecular chaperones capable of delivering proteins such as tumor antigens to antigen-presenting cells (APCs) (e.g., DCs) via specific receptors. This results in the internalization of the HSP complex and presentation of the peptides on class I and class II MHCs.
Recently, HSP-96 has been purified and pulsed on to APCs to provide a personalized polyvalent vaccine in patients with recurrent GBM. Phase I and II trials for recurrent GBM are ongoing. The primary outcome measure in 41 treated patients demonstrated a 6-month OS of 90.2% (95% confidence interval, 75.9–96.8). An interesting additional observation from this study was that patients with lymphocyte counts below the median demonstrated a decreased OS. A phase III randomized trial is now being conducted within the Alliance Consortium (NCT01814813). For this study, patients must have 90% or greater tumor resection to be included. HSP–peptide complex-96 (HSPPC-96) is purified from the resection specimen and used as a vaccine. The primary objective of this trial is to determine whether there is an OS advantage of HSPPC-96 administered with bevacizumab, given concomitantly or at the point of progression, when compared with bevacizumab given alone in patients with resected recurrent GBM.
EGFRvIII: Celldex
EGFRvIII is a tumor-specific mutation of the EGFR that is expressed in 30–35% of primary GBMs and some other common neoplasms. Because the wildtype sequences of human and mouse overlap adjacent to the EGFRvIII mutation, we were previously able to create a murine homologue of this mutation to test its immunogenicity.31 We demonstrated that a peptide vaccine against EGFRvIII induces mutant-specific immunity and prolongs survival in mice bearing EGFRvIII-expressing intracranial tumors.32
Similarly, the EGFRvIII peptide vaccine generates efficacious immune responses in patients. Despite no evidence of EGFRvIII-specific immune responses prior to vaccination, EGFRvIII-specific immune responses were generated and EGFRvIII-expressing cells were eliminated in almost all patients, extending survival without attendant toxicity (OS of vaccinated patients: 26 months; OS of matched controls: 15 months; hazard ratio, 5.3, p = 0.0013).17,33 Moreover, extended OS was predicted by the development of EGFRvIII-specific immune responses. Our EGFRvIII-targeted vaccine is now being tested in an international phase III randomized trial.
Due to the heterogeneous expression of EGFRvIII, however, tumors recurred in nearly all treated patients as a result of outgrowth of EGFRvIII-negative tumor cells.17 Such outgrowth of the EGFRvIII-negative population, however, suggests a significant effect of the vaccine and serves as a lesson regarding the risks of targeting heterogeneously expressed single antigens.
ICT-107
ICT-107 is an intradermally administered vaccine comprised of autologous DC pulsed with six synthetic tumor-associated antigens (TAA): AIM-2, MAGE-1, TRP-2, gp 100, HER-2, and IL-13Rα2. It is currently being tested in a phase II double-blinded, placebo-controlled, multicenter, randomized clinical trial in patients with newly diagnosed GBM, following resection and standard chemoradiation (NCT01280552). The vaccine regimen is given as four induction doses after chemoradiation, then at a maintenance dose until disease progression. The control arm consists of unpulsed DCs. The primary endpoint is OS, defined as the time from randomization until death; the secondary endpoints include PFS, and rates of OS and PFS at 6 months after surgery. The subgroups analyzed in this phase II trial are organized by gender, age, human leukocyte antigen (HLA) type, O6-methylguanine-DNA-methyltransferase (MGMT) status, performance status, and resection status. The latest update at the Society for Neuro-Oncology Annual Meeting in 2014 revealed a promising trend in meaningful clinical benefit for patients treated with ICT-107, providing groundwork for a larger phase III clinical trial (NCT02546102).
DCVax
DCVax-L is a DC vaccine pulsed with patient-specific tumor lysate. It is currently being tested in a phase III placebo-controlled, 2:1 randomized clinical trial in patients with newly diagnosed GBM, who have undergone surgical resection and subsequent chemoradiation (NCT00045968). Crossover is permitted. The study is currently still enrolling. The primary endpoint is PFS and the first secondary endpoint is OS. Additional endpoints include performance status, immune response, and safety.
Optimization: immune monitoring
In recent years, immune monitoring of cancer immunotherapy clinical trials has undergone considerable change. Technical and methodological advances allowing for multiplex profiling of immune phenotypes, definition of regulatory cell subsets, identification of critical signaling molecules, and recognition of biologically important targets and relevant immune endpoints have increased the ability to discover potential immune correlates of clinical efficacy. However, despite this progress, immune correlates of disease-free survival or OS have been difficult to identify. This difficulty has been attributed to: the complexity of host immune system and tumor interactions, the inadequate quality of immune-monitoring assays, delays in the processing and preparation of samples for measuring immune responses, and a failure to measure the immune responses most relevant to cancer progression.
Unexpectedly, there has also been an inability to use T-cell immune response assays to define biomarkers and correlate these with clinical outcomes. This has been attributed to highly variable and often non-reproducible assay results in multicenter trials, as demonstrated by several large international proficiency panels. The use of harmonized assays can reduce this variability and help to build a general framework for assay use in multicenter clinical trials. Researchers from Europe and the USA initiated a project called Minimal Information About T-cell Assays (MIATA),34 which has gained momentum to standardize and harmonize commonly used immune assays such as the enzyme-linked immunosorbent spot assay (ELISpot), MHC tetramer assays, and intracellular cytokine assays. The establishment of universally accepted guidelines for performing and presenting immunological assays in scientific publications will allow the comparison of immune responses across clinical trials. External control of performance in proficiency panels, harmonization guidelines, benchmarks of assay performance, and reference sample repositories and technology will continue to contribute to better-quality immune monitoring.
Immune-monitoring strategies are also continually evolving. Novel assays are being developed to assess signaling pathways in T cells, changes in gene expression and microRNA profiles, as well as the cytokine-induced expression of genes encoding proteins that elicit tumor cell death within tumor and/or stromal cell populations. In addition, standard assays like flow cytometric analysis are being updated to include detection of novel markers – such as the activation markers CD69, HLA-DR proteins and inducible co-stimulator. Immune-monitoring strategies to evaluate immune responses in both peripheral blood and tumor tissues are also being generated. To this end, the concept of “Immunoscore,” initially described for colorectal cancer patients, has been recently introduced as a potentially predictive tool to classify cancers, beside the traditional tumor-staging classification (American Joint Committee on Cancer [AJCC]/UICC [Union for International Cancer Control]-tumor, node, metastasis [TNM]).35 A variety of immunomonitoring strategies commonly utilized today are summarized in Table 5.1.
| Assay | Input | Read-out | Detection method | References |
|---|---|---|---|---|
| Multimer staining | PBMCs; TILs | Antigen-specific T cells | Flow cytometry | 36 |
| ELISpot | PBMCs; TILs | Cytokine-producing antigen-specific cells (colorimetric spot-forming units) | Microscopy | 37 |
| ICS | PBMCs; TILs; whole blood | Polyfunctional T cells; cell surface phenotype markers | Flow cytometry | 38–40 |
| ELISA | Serum or in vitro supernatant | Cytokines, antibodies, growth factors, other proteins of interest | Spectrophotometry | 41,42 |
| CBA | Serum or in vitro supernatant | Bead-bound cytokine analytes | Flow cytometry | 43 |
| MDSC/TAM enumeration | TILs | CD14 and HLA-DR expression | Flow cytometry | 44 |
| Treg enumeration | PBMCs; TILs | FoxP3 (intracellular), CD4, CD25, and CD3 expression | Flow cytometry | 45 |
| Chromium-release | PBMCs/TILs; tumor cells | Cytotoxic immune cells (51Cr decay from compromised tumor cells) | Gamma counter | 46 |
| Flow-based T-cell killing | PBMCs/TILs; tumor cells | LAMP/CD3/CD107 on T cells; 7-AAD on compromised tumor cells | Flow cytometry | 47,48 |
| TCR spectratyping | TILs | T-cell repertoire diversity (frequency of CDR3 region variants in the TCR Vβ family) | RT-PCR | 49 |
CBA, cytometric bead array; ELISA, enzyme-linked immunosorbent assay; ELISpot, enzyme-linked immunosorbent spot assay; HLA-DR, human leukocyte antigen DR; ICS, intracellular cytokine assay; MDSC, myeloid-derived suppressive cell; PBMCs, peripheral blood mononuclear cells; RT-PCR, real-time polymerase chain reaction; TAM, tumor-associated macrophage; Treg, regulatory T cell; TCR, T-cell receptor; TILs, tumor-infiltrating lymphocytes.
Important principles and challenges
The acts of crafting and monitoring immune responses designed specifically as anticancer modalities clearly provide a noteworthy set of challenges, some of which are already highlighted. Yet, within the realm of brain tumor immunotherapy more specifically, there exist unique barriers to success posed by the peculiarities of central nervous system (CNS) immunity, the risks for collateral autoimmune responses, and the uncanny capacity for systemic immune dysregulation exhibited by MG.
Peculiarities: CNS immune privilege?
The CNS has long been considered “immunologically privileged,” but this is an oversimplification of what we now know to be a more complex truth.24 The concept of CNS privilege was derived from early work demonstrating that allographs or xenografts survived longer in the CNS.50 While more current work suggests that the CNS is just immunologically distinct, there remains support for a variety of immune “deficiencies” within the CNS: (1) lower absent MHC expression; (2) lack of resident classic APCs; (3) non-classical lymphatic drainage; and (4) the blood–brain barrier. In the sections below we will consider each of these areas.
Major histocompatibility complex expression
Expression of class I and class II MHC molecules, necessary components to T-cell antigen recognition, remains controversial within the CNS. This is partially due to differential data from laboratories and the use of non-standardized techniques to detect MHC expression. Certainly, cell lines are capable of expressing class I and even class II MHC molecules in vitro. This expression can typically be enhanced by cytokines such as interferon-γ (IFN-γ), but the same effects may not be seen in vivo. Perhaps the more conservative appraisal of the literature suggests that the distribution of class I and II MHC molecules is irregular, under strict regulatory control, and limited to specific cell types. Certainly, the bulk of the evidence suggests that the expression levels are lower.
MHC class I expression in the normal CNS appears to be concentrated in the endothelial and stromal cells, with occasional weak expression in microglia and ependymal cells. The expression level of class I MHC on neurons and glial cells, however, remains unresolved.
MHC class II molecule expression is even more controversial and is generally thought to be limited to select microglial cells and infiltrating DCs.51 There is no consensus regarding the expression level of class II MHC on astrocytes and CNS endothelial cells, although evidence suggests that these cells do express these molecules under pathological conditions, such as multiple sclerosis and experimental autoimmune encephalitis (EAE).52 Such contention perhaps arises due to the true variability of MHC expression across patient samples. Indeed, within 11 glioma samples screened for the presence of class II transactivator mRNA, seven were positive, and the level of expression was highly variable across tumor types. Furthermore, among these samples cultured in vitro, class II expression could be divided into either constitutive or IFN-γ-inducible expression, adding an additional layer of ambiguity.53
The inconsistencies in MHC detection and the sensitivity of its expression to growth and assay conditions make specific determination of CNS MHC levels a particularly difficult issue to resolve within brain tumor immunology. Regardless, however, neither MHC I nor II is expressed at high levels within brain or on glial tumors, posing challenges to T-cell-based immunotherapy.
Antigen-presenting cells
While the CNS certainly has the capability of presenting antigen to the immune system, distinct DCs do not exist primarily in the neural parenchymal microenvironment. Still, microglia, and possibly astrocytes, may be the major or exclusive APCs in the cerebrospinal fluid, and human microglial cells appear to have similar functional and phenotypic characteristics to macrophages and DCs. It has been recently demonstrated that microglia, predominantly located in the perivascular spaces and leptomeninges, are bone marrow-derived and capable of presenting antigen to T cells in vivo.54
Lymphatic drainage
It has been demonstrated clearly that anatomically the brain lacks a conventional lymphatic system. However, it has been well shown that cells and molecules injected into the brain or cerebrospinal fluid can be recovered in the deep cervical lymph nodes in concentrations significantly above plasma levels.55–57 In addition, however, molecules injected into these anatomical spaces can egress through connections between the lymphatic systems and the cranial (olfactory, optic, trigeminal, acoustic) and spinal nerve roots, and the perivascular Virchow–Robin spaces.58 Similar anatomic substrates exist in humans, although these drainage pathways remain of uncertain significance in the development of CNS immune responses.
Blood–brain barrier
The blood–brain barrier exists as a natural physiologic barrier to macromolecules and cells entering the brain. It is not absolute, even under normal conditions, but it has also been well established that the blood–brain barrier is discontinuous in MGs. Debate remains still today as to the degree to which it impedes and/or enhances the efficacy of various therapeutic maneuvers. Most importantly, though, the blood–brain barrier is not a limitation to the passage of activated lymphocytes into the brain, despite lymphocytes in the healthy CNS being a rare finding.59,60 The blood–brain barrier, then, does not appear to be a significant limitation to cell-based immunotherapy. It remains unclear, however, whether the dysregulation of this barrier in the MG setting persists as a greater challenge to passively administered treatments such as targeted antibody therapy.
Collateral autoimmunity: EAE
Autoimmunity within the confines of the CNS must be viewed with greater prejudice than collateral damage arising from therapies targeting more peripheral cancers. Given the consequences of deriving CNS autoimmunity, it likewise becomes important to assess the proclivity for such reactivity with any therapy. It has unfortunately been well documented that fetal and normal brain and systemic antigens are present in human glioma cell lines and brain tissue.61 As a result, vaccination either with human glioma cell lines or GBM tissue risks the induction of an uncontrolled autoimmune response focused on normal CNS or systemic antigens, leading to a syndrome similar to EAE. The susceptibility of humans to such responses was discovered accidentally when patients were vaccinated against rabies using vaccines derived from rabid spinal cord that had been infected with the rabies virus. Since that time, it has been shown that EAE can be readily induced in various species, including non-human primates.62 Bigner et al. vaccinated non-human primates with fresh unfrozen GBM cell suspension in either complete Freund’s adjuvant or incomplete Freund’s adjuvant. In less than 1 month, using either adjuvant, these primates developed severe neurologic symptoms and ultimately irreversible coma and death. Pathologic analysis revealed widespread hemorrhagic necrosis, generalized edema, and widespread perivascular immune infiltration. In addition to these dramatic findings, there should be some concern regarding the induction of EAE based on previous active specific immunotherapy trials in patients.
Immunosuppression in GBM – update and historical perspective
Although immunotherapy can provide more tumor-directed treatment, success will ultimately be linked to our ability to understand and counter the broad host immune defects inherently elicited by MGs. Patients harboring MG possess impaired cell-mediated immunity (CMI) with qualitative, intrinsic T-cell deficits and reduced numbers of circulating lymphocytes. The T cells that do remain are hampered by anergy, IL-2 system dysfunction, TH2-biased responsiveness, decreased NKG2D expression, and inhibition by disproportionate representations of suppressive regulatory T cells (Tregs),63 all products of uniquely potent GBM systemic influences and extrinsic mechanisms for immune escape (reviewed by Dunn et al.).64 T cells that do manage activation and tumor trafficking find themselves faced with equally impressive local and intrinsic means of tumor evasion, including more Tregs, IDO expression, downregulated MHC and B7 family proteins, increased PD-L1, PTEN loss, STAT3 expression/activation, transforming growth factor-β (TGF-β) and IL-10 production, MICA/B secretion, and HLA-E expression, all of which serve to sidestep or directly undermine those immune cells present.65
Early studies
Much of the pioneering work to document CMI impairment in patients with MG occurred in the 1970s and 1980s and is attributable to Thomas Roszman and William Brooks.66 Early studies analyzing host immune responsiveness among patients with benign and malignant intracranial tumors demonstrated reduced delayed hypersensitivity reactions and impaired lymphocyte proliferative responses among patients versus healthy volunteers. A pursuant paper in 197467 repeated the above findings but could not correlate the amount of suppression observed with the degree of malignancy (although a paper by Young et al. in 1976 found that immunocompetence is altered in patients with grade III and IV but not grade I and II tumors).68 However, it was found that the suppression of CMI was lost subsequent to tumor removal, raising the possibility that immunosuppression might become a clinically useful diagnostic tool for recurrence.
In 1977 Mahaley et al.69 comprehensively examined cellular and humoral immune competence in patients with GBM, anaplastic astrocytoma, and meningioma. This paper was the first to examine humoral immunity, and it was one of the first studies to report lymphopenia in brain tumor patients, whereas some of the earlier studies had discussed the presence of normal lymphocyte counts. The group of patients with GBM possessed the smallest number of delayed hypersensitivity reactions responders to two or more antigens and was the only group posting a significant lymphopenia at a preoperative baseline and serial bimonthly postoperative timepoints. Regarding humoral immunity, patients with GBM had mean IgG, IgM, and IgA levels that were consistently within the normal range, although IgM levels experienced a decline over time after an initial elevation. Examination of tetanus and influenza antibody titers demonstrated serum tetanus antibody responded to boost, but serum influenza antibody eventually declined progressively, despite boosting.
Studies of patients with MG continued to document depressed cutaneous reactivity to common antigens, as well as the ability of patient plasma to inhibit lymphocyte responsiveness in mixed lymphocyte reaction or to mitogens such as is phytohemagglutinin (PHA). Additional work70 confirmed the presence of lymphopenia in patients with glial tumors and correlated the severity of lymphopenia with tumor grade such that patients with GBM presented with a 37% reduction in the number of circulating lymphocytes. Furthermore, the lymphopenia was characterized by a selective depletion of T cells, while B-cell levels remained normal (when corrected for the lymphopenia). In 1976, Young et al. observed that lymphocytes from approximately 50% of patients with GBM were significantly inhibited in their response to either PHA or concanavalin A, even when cultured in the presence of normal plasma.68 These data raised the novel possibility that defects existed which were intrinsic to the lymphocyte compartment.
By 1980, it was clear that circulating humoral factors clearly could not account for the impaired CMI observed in patients with primary brain tumors, as patient T-cell dysfunction persisted despite culture in normal human sera.71 Importantly, the existence of T-cell lymphopenia could not explain these observations, as purified T-cell populations provided similar results. Neither increasing the number of lymphocytes in culture nor altering the duration of the culture period corrected the observed proliferative defects. Thus, the first conclusive evidence was offered for alterations in the functional qualities of lymphocytes from patients with glioma, bringing T-cell dysfunction to the center stage.
Lymphocyte dysfunction in patients with malignant glioma
T-cell dysfunction has been progressively narrowed to the helper T-cell subset, as T cells from patients proved unable to provide adequate helper activity in the pokeweed mitogen (PWM) response. Additionally, it was found that CD4+ T cells were significantly reduced among patient peripheral blood leukocytes (PBL: 40% vs. 55% for controls), while the proportion of CD8+ T cells remained normal.72 Furthermore, patient-derived CD4+ T cells proliferated less well in response to mitogen stimulation than CD8+ T cells obtained from the same patient or CD4+ T cells obtained from healthy controls.73
While allogeneic PWM cultures were being used to localize defects to within the CD4+ subset of T cells, cytokinetic analysis of PHA-induced [3H]-thymidine incorporation indicated defects in the ability of PBL from patients with brain tumor patients to undergo clonal expansion. Measurements of indirect IL-2 activity in culture supernatants also demonstrated significantly lower levels of the cytokine in cultures containing patient cells; this has proved to be a crucial observation in the work to gain understanding of the mechanisms behind T-cell failures. Furthermore, the addition of exogenous IL-2 was unable to reverse the existing proliferative defects,74,75 which suggested that IL-2 receptor (IL-2R) signaling or expression might also be defective. One of the earliest works in this regard examined the percentage of IL-2R+ lymphocytes at various timepoints after PHA stimulation and found that this number was indeed reduced when compared to normal values.76
Examination of T-cell anergy among patients with MG has revealed that T-cell receptor (TCR)-mediated signaling is defective as well. A 1997 study by Morford et al.77 demonstrated both PHA and anti-CD3 monoclonal antibody-stimulated patient PBL and T cells exhibited marked defects in early transmembrane signaling events, as measured by the tyrosine phosphorylation of a variety of proteins, including decreased phosphorylation of pp100 and phospholipase Cγ1 (PLCγ1). Additionally, PLCγ1 and p56lck protein levels were reduced in patient T cells, and the cells mobilized less calcium after stimulation. Proliferative capacity was not restored following stimulations with phorbol myristate acetate and ionomycin, indicating that that observed anergy in patient T cells was attributable to defects in early transmembrane signaling events associated with TCR / CD3 stimulation.
Cytokine dysregulation
There are a number of lines of evidence that gliomas can elicit a shift in patients from Th1- to Th2-type cytokine production. Most prominent, perhaps, are the production and secretion by the tumor itself of TGF-β isoforms and other molecules such as IL-10 and prostaglandin E2.78–81 Human TGF-β1 and –β2 have been isolated from GBM supernatants,79–81 and these cytokines are known to suppress the IL-2-dependent generation of CTL from PBL and tumor-infiltrating lymphocytes (TIL); to inhibit IL-2R expression on T cells; to reduce IL-1- and IL-2-dependent proliferation of T and B lymphocytes; to depress the cytotoxicity of natural killer (NK) cells and their activation by IFN-γ; to reduce IFN-γ production; to downregulate MHC class II expression and presentation; to suppress Th1 cytokine synthesis; to inhibit the accessory function and antigen-presenting capacity of monocyte/macrophages, and to suppress the production of numerous proinflammatory cytokines, including tumor necrosis factor (TNF)-α, IL-1, IL-6, and IL-8 by monocytes following activation (reviewed by Li and Flavell).82 The potential for immunosuppressive factors, such as TGF-β, to abolish a cell-mediated antitumor immune response has already been demonstrated experimentally.83 IL-10 inhibits the synthesis of IFN-γ, IL-1α, IL-1β, IL-6, IL-8, and GM-CSF by activated monocytes and lymphocytes.84 It also possesses the capacity to reduce the antigen-presenting function of monocytes, thereby hindering antigen-specific T-cell proliferation. Prostaglandin E2, at high concentrations, can impact T-cell activation, but the relevance of the levels secreted by gliomas is questionable.
In addition to the influence of tumor itself, studies demonstrated evidence for a shift to Th2 responses in patient cellular immune responses as well. Patient PBL failed to secrete the Th1 cytokine IFN-γ, particularly in response to Candida mannoprotein.74,84 This failure, however, could be a byproduct of the intrinsic dysfunction of CD4+ T cells, which are the major producers of IFN-γ. PBL from patients with glioma also secrete increased levels of IL-10, while failing to elaborate IL-12 (both the p40 and p70 subunits) in response to stimulation with Staphylococcus aureus Cowan strain.85 The same cytokine dysregulation holds true for TILs isolated from fresh glioma samples. One study employing semi-quantitative reverse transcriptase polymerase chain reaction demonstrated TILs isolated from glioma patients predominantly expressed IL-4 and GM-CSF and largely did not express IFN-γ, IL-2, or TNF-β at levels higher than normal PBL.86 Most recently, evidence has surfaced that gliomas may directly negatively regulate T-cell cytokine production and proliferation via cell surface expression of programmed death receptor ligand 1 (PD-L1, also called B7-H1), which would interact with PD-1 on tumor-specific T cells and potentially permit tumor escape from the host immune system.87
Regulatory T cells
As purified T-cell populations from patients with glioma were unresponsive, and as defects intrinsic to the T-cell compartment were at least partially responsible for the impaired CMI, it was suggested that the “results might indicate the expansion of an otherwise normally present subpopulation of cells which…are capable of modulating the responsiveness of other lymphocytes.”71 The existence of such suppressor cells had been proposed in 1971,88 and others had shown them to be maximally responsive at suboptimal concentrations of mitogen, a notion consistent with the observed pattern of mitogenic responsiveness for patient lymphocytes at the time. Unfortunately, lymphocyte-mediated suppressor activity proved difficult to reproduce,72,89 and the concept lost favor. However, the last 15–20 years brought a paradigm shift to modern immunology through uncovering immunoregulatory cell activity focused in the CD25+ (IL-2Rα+) fraction of CD4+ T cells. The now recognized “regulatory T cells” (Tregs) represent the CD25+FOXP3+ subset of CD4+ T cells, which equates to 5–10% of CD4+ T cells in the blood of both mice and humans. Their physiologic function, following selection in the thymus for their moderate degree of self-avidity, appears to be attenuation of pathologic autoimmune responses by recognition of tissue-specific self-antigens in the periphery. They potently inhibit CD4+ and CD8+ T-cell activation and proliferative responses, downregulating production of IL-2 and IFN-γ in favor of the Th2 cytokines frequently encountered in MG: IL-10 and TGF-β. These two cytokines propagate the tolerizing effects and can even confer a regulatory phenotype upon targeted T cells (Figure 5.1).
Tregs have been found at elevated levels in the tumors and/or peripheral blood of patients with a variety of cancers, and demonstrably restrict tumor-specific immunity. As their activity mimics much of the defective CMI observed in MG, Tregs have increasingly been implicated in the muted immune responses observed in glioma patients. Furthermore, in the blood of patients with MG the Treg compartment is a disproportionately large fraction of the reduced CD4 compartment, a homeostatic disruption that solicits many of the long-recognized systemic T-cell deficits.63 Similar Treg increases occur locally at the tumor with demonstrated implications for the antitumor immune response.90 Importantly, patient T-cell deficits prove reversible with Treg depletion,63,91 an in vivo intervention in mice that confers prolonged survival.63,92,93 Taken together, these observations point strongly to a significant role for Tregs as one of the crucial missing links in our understanding of the cell-mediated immune defects observed in glioma patients over the last few decades.
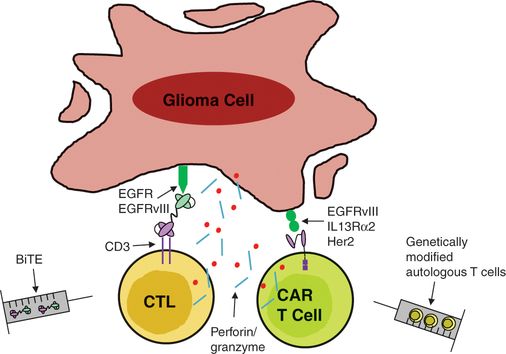
T-cell-based approaches to glioma immunotherapy. There exist several engineered methods for directly targeting effector T cells to glioma cells. Bispecific T-cell engagers (BiTEs) are generated by linking the variable region of an anti-TAA antibody to the variable region of an anti-CD3 antibody. Additionally, autologous T cells can be genetically modified to express a chimeric antigen receptor (CAR), consisting of the variable region of an anti-TAA antibody fused to intracellular T-cell signaling molecules. Such strategies induce effector T-cell function upon binding to the tumor cell and eliminate the necessity for antigen recognition in the context of major histocompatibility complex molecules on the tumor.
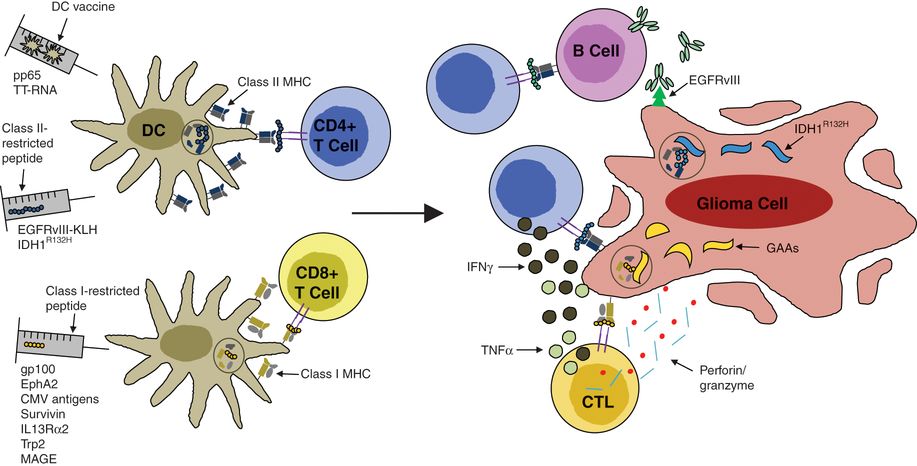
Peptide and dendritic cell vaccines for glioma immunotherapy. Vaccines can be delivered in the form of short peptide epitopes or through ex vivo generation of autologous antigen-presenting cells (dendritic cells) pulsed with TAAs. Such strategies allow for priming of antigen-specific T cells and their reactivation upon recognition of major histocompatibility complex (MHC)/peptide complexes presented by glioma cells. Such epitopes are known to be either MHC class I or class II restricted, giving rise to different antitumor effector mechanisms.
A black and white version of this figure will appear in some formats. For the color version, please refer to the plate section.
Emerging topics
Immunotherapy remains one of the most rapidly expanding arenas within cancer research and treatment. In an era when focus is trending towards notions of personalized and custom therapeutics, immune-based treatments present themselves as a vital component to multifaceted antitumor approaches. As we strive for better potency, acceptable safety, and evasion of tumor countermeasures, new discoveries and trends emerge that include myeloconditioning / lymphodepletion, immune checkpoints, genetically modified T cells, antibodies with dual specificity, and new antigenic targets, to name a few. A brief introduction to these topics is offered here.
Lymphodepletion
One of the largest lessons learned in the ALT arena has been the antitumor benefit of myeloconditioning prior to adoptive transfer.94 Lymphodepletion using total body irradiation or even non-therapeutic chemotherapy in mice and humans has been applied to optimize immunotherapy by leveraging the induction of homeostatic cytokines and the consequent T-cell proliferation.95 In fact, this treatment has been shown to induce massive T-cell proliferation and significant amplification of tumor-specific immune responses.95 Lymphocytes specific for antigens that predominate during this recovery period, like those provided in the form of a vaccine, have a competitive advantage and become disproportionately over-represented in the recovering lymphocyte population.96,97
In the case of GBM, standard of care temozolomide (TMZ), a potent methylating agent, also induces lymphodepletion, which theoretically could eradicate the production of an immune response, especially in the setting of serial cycles. However, our published preclinical data demonstrate that, when the vaccine is timed during homeostatic T-cell recovery after TMZ-induced nadirs, TMZ administration synergizes with vaccination and adoptive T-cell therapies by increasing levels of circulating homeostatic cytokines.94 These effects are dose-dependent, with enhanced efficacy observed at the highest doses of TMZ, presumably due to a greater degree of host lymphodepletion.17,98 We have also demonstrated that EGFRvIII-specific immune responses induced in patients with GBM by Rhindopepimut vaccination can be maintained in the context of serial cycles of therapeutic TMZ.17,98
Interestingly, lymphodepletion may also permit specific eradication of Tregs. Tregs are known to be uniquely dependent on the high-affinity IL-2 receptor (IL-2Rα)(CD25) for their function and survival, and defects in IL-2Rα in mice and humans lead to multi-organ inflammatory infiltrates. Monoclonal antibodies that block IL-2Rα abrogate Treg function in animal models, but can also inhibit effective antitumor immune responses in mice and humans with autoimmune disorders and organ transplants.93 This negative impact on activated immune responses is likely due to a requirement for IL-2Rα signaling by effector T cells or a recently described indirect elimination of effector T cells by regulatory NK cells activated by IL-2Rα-specific monoclonal antibodies. Our preliminary data demonstrate that αnti-IL-2Rα monoclonal antibodies have very different effects on activated immune responses in mice undergoing homeostatic proliferation in response to transient lymphodepletion, as might be seen after therapeutic cycles of cytotoxic chemotherapies like TMZ. In the context of lymphodepletion-induced homeostatic γ common cytokines, IL-2 may be dispensable for effector T cells while Tregs remain absolutely dependent on it. These findings will be broadly applicable to cancer immunotherapy and basic immunology.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


