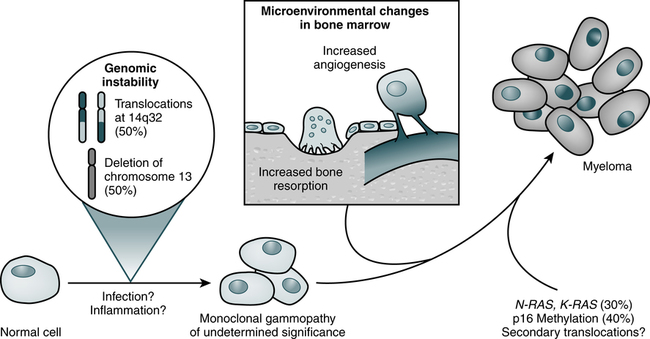
At the conclusion of this chapter, the reader should be able to: • Compare the general characteristics of monoclonal and polyclonal gammopathies. • Describe and compare the etiology, epidemiology, signs and symptoms, immunologic manifestations, diagnostic evaluation, and treatment of multiple myeloma and Waldenström’s primary macroglobulinemia. • Explain and contrast the characteristics of other monoclonal disorders, such as monoclonal gammopathy of unknown significance. • Analyze a case study related to immunoproliferation. • Correctly answer case study related multiple choice questions. • Be prepared to participate in a discussion of case study related critical thinking questions. • Describe the principle and application of the Bence Jones Protein Screening Procedure. Myeloma cells proliferate slowly in the marrow (Fig. 27-1; Table 27-1). Less than 1% divide at any one time and myeloma cells do not differentiate. The absolute number of these cells correlates with disease activity and predicts the progression of disease in smoldering multiple myeloma. Circulating myeloma cells may disseminate the tumor within the bone marrow and elsewhere. Table 27-1 Three Phases of Disease Progression in Multiple Myeloma ∗Growth fraction is the rate of atypical cells proliferating in the bone marrow. From Bataille R, Harousseau JL: Medical progress: multiple myeloma, N Engl J Med 336:1657–1664, 1997. IgG myeloma is the most common form of MM (Table 27-2). Four subtypes of IgG heavy chains are known to exist among patients with IgG myeloma. Cases of IgG myeloma are distributed as follows: 65% are gamma G1, 23% gamma G2, 8% gamma G3, and 4% gamma G4 subclass. The only subclass-dependent difference is the greater propensity for patients with IgG3 myeloma to experience hyperviscosity syndrome, similar to the manifestation in WM. Table 27-2 Distribution of Immunoglobulin Types in Patients With Multiple Myeloma About 90% of patients with MM have broadly disseminated destruction of the skeleton, which is responsible for the predominance of bone pain. These abnormalities consist of punched-out lytic areas (Fig. 27-2), osteoporosis, and fractures in about 80% of patients. The vertebrae, skull, thoracic cage, pelvis, and proximal humeri and femurs are the most frequent sites of involvement.
Immunoproliferative Disorders
Multiple Myeloma
Pathophysiology
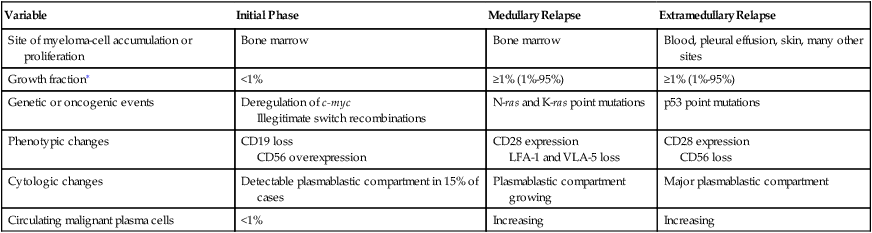
Variable
Initial Phase
Medullary Relapse
Extramedullary Relapse
Site of myeloma-cell accumulation or proliferation
Bone marrow
Bone marrow
Blood, pleural effusion, skin, many other sites
Growth fraction∗
<1%
≥1% (1%-95%)
≥1% (1%-95%)
Genetic or oncogenic events
Deregulation of c-myc
Illegitimate switch recombinations
N-ras and K-ras point mutations
p53 point mutations
Phenotypic changes
CD19 loss
CD56 overexpression
CD28 expression
LFA-1 and VLA-5 loss
CD28 expression
CD56 loss
Cytologic changes
Detectable plasmablastic compartment in 15% of cases
Plasmablastic compartment growing
Major plasmablastic compartment
Circulating malignant plasma cells
<1%
Increasing
Increasing
Epidemiology
Type of Protein
Multiple Myeloma (%)
IgM
12
IgG
52
IgA
22
IgD
2
IgE
Rare
Light chains (kappa or lambda)
11
Heavy chains
Rare
Monoclonal proteins
<1
Nonsecretory myeloma
1
Signs and Symptoms
Skeletal Abnormalities
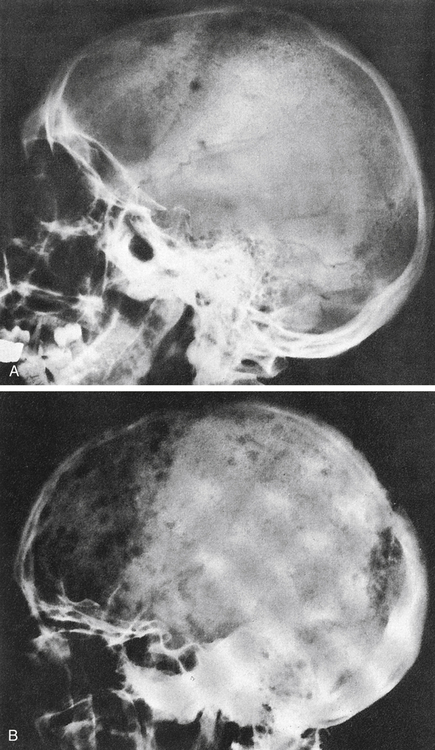
A, Several scattered, small, well-marginated lytic lesions appear in calvarium, located in normally mineralized bone. Multiple lytic lesions can also be seen in the mandible. B, Multiple circumscribed lytic lesions crowd bones throughout skull. Lesions are still discrete and margins of most are fairly sharp. (From Newton TH, Potts DG: Radiology of the skull and brain, St Louis, 1971, Mosby.)
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Oncohema Key
Fastest Oncology & Hematology Insight Engine