Fig. 18.1
Polarization of tumor-associated macrophages in glioma. Notice the distinct M1 and M2 phenotypes (Reprinted from Li and Graeber [18], with permission)
Alternatively activated M2-type TAMs are the predominant immune cell type in malignant glioma, and their presence has been shown to correlate with histological grade [46]. A recent investigation revealed increased expression of the M2 markers CD163 and CD204 by TAMs in WHO grade IV gliomas, compared to WHO grades II and III gliomas [47]. The perverse polarization of TAM precursors, both resident microglia and peripheral derived monocytes, to the alternative M2 state is generally believed to occur as these cells encounter the myriad cytokines, growth factors, and surface antigens of the tumor microenvironment. Among the factors implicated in the active recruitment and altered polarization of monocytes by CNS tumor cells, monocyte chemoattractant proteins 1 (MCP-1/CCL-2) and monocyte colony-stimulating factor (M-CSF) are believed to drive local recruitment and proliferation of TAM precursors, while TGF-β, IL-4, IL-10, and IL-13 together orchestrate polarization to the alternative M2 phenotype [47, 48]. Importantly, this polarization toward a M2 TAM phenotype takes place in the absence of IFN-γ, a potent driver of the classical M1 phenotype [49].
The absence of IFN-γ is likely due to the suppression of its principle source, activated type 1 T-helper cells (discussed below).
18.4.4 Myeloid-Derived Suppressor Cells
Recent refinements of the M1/M2 TAM characterization scheme describe a more heterogeneous population of systemically distributed M2 TAM-like myeloid-derived immunosuppressive cells at intermediate stages of maturation, which are able to suppress multiple phases of the immune response [50]. These myeloid-derived suppressor cells (MDSCs) have been shown both to perpetuate tumor-promoting microenvironments and to distribute peripherally to hinder lymphocyte activation in immune organs. MDSCs are therefore implicated in the general systemic immunosuppression observed in patients with malignant gliomas [48]. Recent evidence suggests that MDSC precursors must be exposed to the concentrated cocktail of immunomodulatory mediators and cell-cell interactions in the tumor microenvironment to become MDSCs [48]. These observations suggest that naïve monocyte traffic to the tumor microenvironment, mature into immunosuppressive MDSCs, and then redistribute systemically [48]. Systemically circulating MDSCs present a poorly understood hurdle to remediating CNS cancer immune suppression. Their heterogeneous expression profile and systemic distribution allow a potentially broad and widespread armament of immunosuppressive functions. If indeed these cells are generated by local tumor-derived factors of the microenvironment, as in the more clearly defined M2 TAM phenotype, then disabling the local “monocyte-educating” mechanisms of tumor cells may reduce the generation of MDSC.
18.4.5 Lymphocytes and Regulatory T cells
Lymphocyte effector cells are major players in antineoplastic immunity, yet lymphocytes which traffic to cancers of the CNS are disabled, reprogrammed to immunosuppressive phenotypes, and subsequently permitted to remain within tumor through failure of natural anergic cell deletion. As discussed above, the process of T-cell activation by APCs is severely hindered in CNS cancers by reductions in MHC and co-stimulatory molecules on both tumor cells and surrounding APCs and by the milieu of T-cell-deactivating mediators within the tumor microenvironment. NK cells are known to initiate deletion of T cells with reduced expression of MHC or co-stimulatory molecules, releasing TNF-α and IFN-γ (reviewed in [50]). This fail-safe mechanism is believed to be disabled by the immunosuppressive milieu of the local tumor microenvironment, most notably by IL-10, and by activation of the NK cell inhibitory receptor KIR2DL through the ligand HLA-G, which is expressed on Tregs [51]. Through these mechanisms, T lymphocytes that are polarized to immunosuppressive phenotypes are permitted to remain within CNS tumors.
Ongoing research implicates Tregs as a major lymphocyte player in CNS tumor immune biology. An increased systemic prevalence of Tregs among T cells has been observed in malignant glioma, consistent with their role in suppressing the immune rejection of neoplastic cells [52]. In addition, Treg infiltration of brain tumors has also been demonstrated, and in the case of gliomas, the fraction of Treg correlates with tumor grade [52, 53]. These observations reflect the significant role Tregs play as a negative immune modulator of lymphocytes both within the tumor and peripherally in lymphoid organs, leading to immune evasion by tumor cells.
Investigation of the origin, recruitment, expansion, and immunomodulatory effect of Tregs in malignant gliomas is an active effort within the tumor immunology field. Recent evidence shows that T cells may be converted to CD4+/Foxp3+-induced Tregs (iTregs) peripherally through exposure to APCs or suboptimal TCR stimulation in the presence of high levels of TGF-β, as is present in the tumor microenvironment [40].
Both iTregs and thymus-derived natural Tregs (nTregs) have been shown to infiltrate and proliferate within CNS tumors. These cells migrate in response to tumor-secreted MCP-1, which binds CCR4, a receptor highly expressed on Tregs and their precursors [51]. The mechanisms by which Tregs elicit immunosuppression involve Foxp3-mediated expression of the immunosuppressive cell surface ligands glucocorticoid-induced tumor necrosis factor receptor (GITR), cytotoxic T-lymphocyte antigen (CTLA-4), and human leukocyte antigen G (HLA-G), as well as through the contribution of immunosuppressive cytokines TGF-β and IL-10 into the microenvironment [54]. This inhibitory signal replaces the stimulatory interaction between T-cell protein CD28 and APC co-stimulatory molecules B7-1 and B7-2 to prevent activation. HLA-G on placental cells has been shown to contribute immune tolerance in pregnancy by binding the KIR2DL receptor of NK cells, blocking activation in the presence of cells lacking MHC or co-stimulatory molecules. By this mechanism, Tregs are hypothesized to disable NK cell surveillance.
18.5 STAT3 Pathway
As discussed, many soluble mediators and cell surface molecules expressed by tumor cells, TAMs, and Tregs participate to establish a potently immune-disabling microenvironment. Expression profiles across these various cellular players are similar, raising suspicion for unifying mediators of signal transduction or gene expression common to these shared phenotypes. Signal transducer and activator of transcription protein 3 (STAT3), a transcription factor active in both glioma cells and TAMs, has been shown to influence multiple immunosuppressive signaling pathways implicated in CNS tumor-induced immunosuppression [55].
Furthermore, considering the myriad targets of STAT3 modulation, activation of this intracellular mediator may also augment CNS tumor angiogenesis and stromal remodeling [56]. STAT3 activation in glioma TAMs is induced downstream of many mediators known to constitute the local microenvironment such as IL-10, IL-6, EGF, and FGF [57]. In both tumor cells and TAMs, STAT3 decreases the expression of surface molecules necessary for antigen presentation such as MHC II, B7-1, and B7-2 and upregulates M2-specific immunomodulatory mediators including IL-10, EGF, VEGF, and various matrix metalloproteinases (MMPs) (reviewed in [18]). Experiments blocking the activation of STAT3 in gCSCs cocultured with allogeneic T-cell precursors demonstrate reduced Treg differentiation and reduced overall T-cell apoptosis [58]. Therefore, STAT3 may serve as a critical “molecular hub” linking multiple immunosuppressive pathways in CNS tumor cells and M2 TAMs. STAT3 target molecules such as IL-10 and IL-6 have been shown to subsequently trigger STAT3 activation [59], leading authors to propose a feedforward mechanism of reinforced STAT3 activation, which may account for its constitutive activation in both glioma cells and glioma-infiltrating TAMs.
18.6 Cytomegalovirus in Glioma
Accumulating evidence demonstrating an association between active human CMV infection and malignant glioma has inspired exciting innovations to current treatment strategies. A recent investigation reported the presence of CMV-associated nucleic acids and proteins in over 90 % of ex vivo GBM specimens analyzed. Neither HCMV-associated nucleic acids nor proteins were present in surrounding normal brain specimens, and over 80 % of recently diagnosed GBM patients also demonstrated CMV DNA in peripheral blood samples [60]. Though CMV is known to infect 50–80 % of the American population, effective immune control typically limits active disease to the immunosuppressed [61]. It remains unclear if the high prevalence of active CMV infection in glioma patients plays any role in tumor pathogenesis or if tumor growth simply provides an environment permissive of local reactivation and propagation of the virus. Regardless, the presence of CMV in these tumors may be important considering its known potential to modulate growth, invasiveness, and immunological recognition of infected cells (reviewed in [62]). Indeed, active CMV infection has been shown in astrocytes to reduce expression of molecules necessary for antigen presentation, increase the expression of TGF-β and IL-10, and limit the susceptibility of infected cells to apoptotic pathways [63, 64]. Elucidation of the impact CMV virus has on the immunosuppressive phenotypes of CNS tumor cells will require extensive investigation. The presence of viral antigens specifically in tumor cells may allow for tumor cell-specific targeting through the use of CMV antigens in CNS tumor vaccines. If in fact active CMV activation contributes to cellular transformation or malignant behavior, then vaccination strategies against its antigens could additionally provide a functionally disabling therapy toward preventing recurrence.
18.7 Immunoediting in CNS Cancer
As most human CNS tumor analysis is conducted on ex vivo specimens acquired from surgical excision following presentation of clinical deficits, the data and conclusions may not be representative of earlier stages of immune system and tumor interaction. Thus, whereas it is possible to study the immunosuppressive environment present in a malignant tumor, the sequence of events that leads to this state remains obscure. The theory of tumor immunoediting has emerged as a paradigm for understanding the dynamics of tumor progression and immunosuppression. The hypothesis proposes three distinct phases: an initial elimination, a period of equilibrium, and, finally, cancer cell immune escape [65] (tumor immunoediting is summarized in Fig. 18.2). Due to genetic instability and rapid proliferation, tumor cells are generated with different immunogenic antigens in a developing tumor. In the initial elimination phase, cytotoxic immune cells target and eliminate those cancer cells that are highly recognizable and lack immune-evasion mechanisms, leading to the selection of poorly immunogenic and/or immunosuppressive tumor cells. Elimination is limited, and some tumor cells are not eradicated, either due to their antigenic or immunosuppressive-related gene expression profile, allowing these cells to survive the initial immune surveillance and enter an equilibrium phase. In this phase, there is a dynamic balance between the antitumoral immunity and tumor cell expansion. During this long phase, there is no clinical tumor burden. The prolonged latency period during equilibrium is thought to constitute an editing state in which neoplastic cells that are susceptible to the host immunity are eradicated, and those that are not recognized are selected to survive. Finally, the escape phase occurs when those tumor cells that are not detectable or have developed mechanisms to avoid immune recognition are selected and grow into a symptomatic lesion (Fig. 18.2).
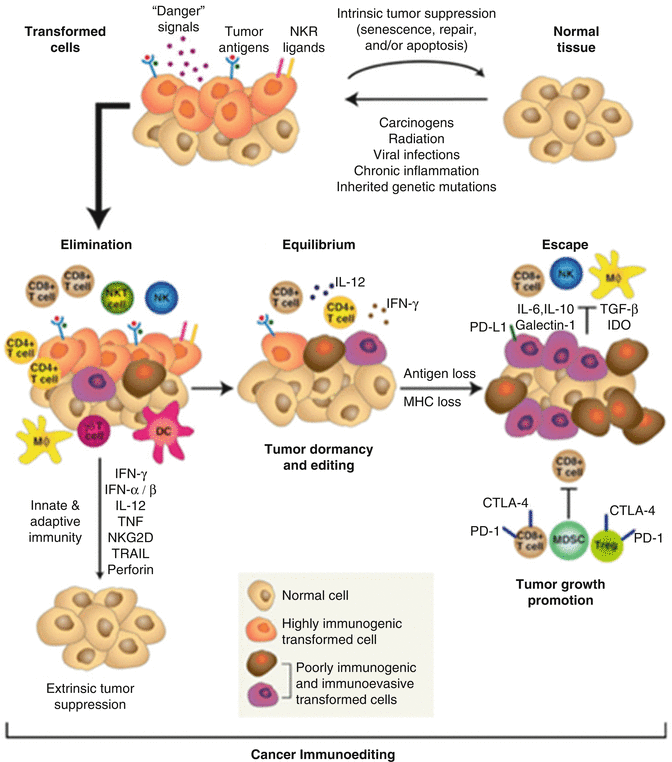
Fig. 18.2
Cancer immunoediting paradigm, highlighting the three proposed phases of immunoediting: elimination, equilibrium, and escape (Reprinted from Schreiber et al. [65], with permission of AAAS)
Considering the competence of immune surveillance and activation within the CNS, the principles of tumor immunoediting are believed to apply to CNS cancers. Support for the paradigm of immunoediting in CNS cancers comes from few transplant studies, citing the transmission of glioma tumors from liver and kidney organ donors to transplant recipients and from observations in ongoing immunotherapy trials. The first report of this phenomenon involved a 44-year-old woman with primary biliary cirrhosis who received an orthotopic liver transplant from a 14-year-old brain-dead donor with a glial tumor that had infiltrated the pons, pituitary, and spinal cord. Following 9 months of immunosuppression, the recipient developed several liver lesions that appeared histopathologically similar to that of the donor’s glial tumor, suggesting immune escape of glioma cells maintained in quiescent immune equilibrium prior to transplantation [65].
A similar report documented two recipients who each received a kidney from a deceased donor with GBM. Both recipients developed renal masses after approximately 18 months, which upon organ removal were pathologically consistent with GBM [66]. Further evidence comes from current GBM vaccine trials (detailed below). Analysis of recurrent GBM specimens following use of a vaccine targeting the highly expressed variant EGFRvIII in GBM demonstrated a paucity of EGFRvIII expression, suggesting successful elimination of the EGFRvIII-expressing cells, followed by equilibrium and subsequent escape of cancer cell subpopulations which did not express EGFRvIII [67]. Ongoing investigation of the dynamic interactions between immune cells and tumor cells throughout the multiphasic progression of CNS tumors will test this theory of immunoediting in CNS cancers and potentially elucidate opportunities to enhance elimination and redirect the eventual failure of equilibrium.
18.8 Immunotherapy
In general terms, the CNS tumor immunotherapy strategies are focused on two goals: to direct the recognition of CNS cancer cells by immune effector cells necessary for a tumoricidal response and to counteract tumor-derived immunosuppression, thus leading to an effective antitumor activation state. A growing appreciation of the necessity for multimodal immune modulation in achieving durable control of CNS tumors through immune-based therapy has led to the combination of both strategies in preclinical and early clinical trials.
In efforts to enhance tumor detection by the immune system, antigen-specific vaccinations and primed dendritic cell-based infusions have both demonstrated promising results. With regard to efforts aimed at disabling immunosuppressive mechanisms, those targeting Tregs and immunomodulatory cytokines have shown preliminary success.
Some authors argue that surgery offers a means for disabling tumor-related immunosuppression by removing the bulk of immunosuppressive cells and mediators within the tumor [44]. Additionally, elimination of the mass effect and edema caused by a large tumor allows for discontinuation of steroids, which confer an iatrogenic immunosuppressive state to the patient. An example of the benefit of resection in the context of immunotherapy has been shown in post-resection GBM patients who, without a significant tumor mass and actively progressing disease, responded better to dendritic cell-based vaccines than did those who had received biopsy alone [68]. For this reason, many recent clinical trials of immune-based therapy in GBM patients are focused on patients who first receive a surgical resection of their tumor.
18.8.1 Adoptive Therapy
Considering the potent tumoricidal properties of activated lymphocyte effector cells, an obvious strategy toward overcoming the in vivo hindrances to adaptive immune activation utilizes infusion of in vitro activated autologous lymphocytes back into patients. Lymphokine-activated killer (LAK) cells are populations of autologous peripheral lymphocytes that can be reinfused into tumor-bearing hosts either peripherally or intraoperatively into post-resection surgical cavities following in vitro culture in the presence of IL-2 [69]. Multiple phase I clinical trials have investigated LAKs in patients with high-grade gliomas and medulloblastomas (reviewed in [70, 71]. The most promising of these trials included 40 GBM patients treated with intratumoral LAKs and demonstrated a slight but significant increase in median survival in the absence of any toxicity [69]. Unfortunately, additional trials could not reproduce these effects and were further hindered by variant levels of toxicity as the reinfused LAKs demonstrated cytotoxic properties that were not specific to tumor cells. Lower cellular doses of intralesional LAK are under continued investigation as adjuvant treatment of GBM [69].
An extension of LAK strategies to direct more tumor-specific targeting involved the collection of lymphocytes from the lymph nodes or peripheral blood of patients with CNS tumors after peripheral injection of irradiated autologous tumor cells (ATCs) and granulocyte/macrophage colony-stimulating factor (GM-CSF). The harvested lymphocytes were then stimulated in vitro with IL-2 and subsequently reintroduced into the tumor-bearing host [72]. Variations in this scheme include additional ex vivo exposure to ATCs [72] or tumor-infiltrating lymphocytes isolated from resection specimens [19] during in vitro stimulation. Despite reduced toxicity and more objective tumor-specific targeting as compared to LAKs stimulated with IL-2 alone, effects on clinical outcome were minimal across these trials [19, 73].
18.8.2 Vaccination Strategies
Cancer vaccination strategies utilize tumor antigen-driven stimulation of host immune processes to target transformed cells. Cancer vaccines are designed to direct tumor-specific cellular immunity by stimulating the proliferation of high-avidity T cells capable of homing to and selectively attacking transformed cells within a tumor. Some of the major challenges to this strategy include failure of the delivered stimulus to adequately activate T cells, relative lack of tumor-specific antigens that are expressed by a large fraction of tumor cells, nonspecific targeting by stimulated T cells of healthy bystander cells resulting in toxic autoimmunity, and disabling of activated tumor cell-specific T cells by the local microenvironment. To overcome these issues, some vaccination strategies utilize reinfusion of autologous tumor material following ex vivo manipulation [74], as well as the use of non-antigen-specific tumor lysate preparations [75]. More recently, purified tumor antigen formulations have also been attempted as direct peptide infusions and as a priming stimulus to DCs prior to their infusion.
18.8.2.1 Autologous Tumor Material
ATCs may be harvested from ex vivo tumor resection specimens and used to generate direct CNS vaccination formulations. Subcutaneous or intradermal injection of autologous tumor material is believed to circumvent the immune-disabling tumor microenvironment by providing specific immune-stimulating material to peripheral DCs. Prior to their use in vaccination strategies, this tissue is processed to isolate whole tumor cells, parts of cells, or simply protein extracts and often inactivated by radiation or genetic modification. Eight trials have employed such strategies to treat GBM, including one phase I clinical trial [74], two case reports [76, 77], and five pilot vaccination studies (reviewed in [70]). In three of the pilot studies, processed cells were delivered concomitantly with adjuvant compounds, including IL-2 [76], IL-4 [77], and B7-2 plus GM-CSF infusions [78]; the amount of cells delivered varied across trials.
The induction of an immune response was demonstrated in over half the patients enrolled in each trial, with evidence both in peripheral blood [79] and at the tumor site [80]. Toxicity was minimal and no patient demonstrated severe adverse effects. Furthermore, clinical benefit was demonstrated with nearly 50 % overall survival across five studies, which recorded three complete responses, four partial responses, two minor responses, and six cases of stable disease in 48 total GBM patients [74, 76–78]. The phase I clinical trial of ATC vaccination included a concomitant infusion of GM-CSF through a programmable pump and effected a significant increase in survival in three of the five patients who demonstrated a postvaccination immune response, out of a total of nine who were treated [74].
18.8.2.2 Dendritic Cell-Based Vaccination Strategies
As discussed, activation of T cells that specifically target brain tumor cells is limited by a reduction in the expression of molecules necessary for effective antigen presentation, including MHC class I/II and co-stimulatory molecules [28]. To overcome this limitation, DCs from patients with malignant brain tumors may be extracted, activated in vitro with tumor-derived antigens favoring APC maturation, and reintroduced as potent activators of tumor-specific T cells. This approach can lead to the generation of tumor-specific T-helper cells (Th) capable of altering the composition of the microenvironment through expression of immune-activating mediators and, subsequently, the activation of CTLs and NK cells capable of selectively eliminating tumor cells. Furthermore, the generation of memory T cells following introduction of tumor antigen-primed DCs presents the potential for lasting immunity to counter the recurrent proliferation of residual cancer cells. Indeed, coculture of glioma-associated antigen-primed DCs with undifferentiated lymphocytes has been shown to induce activation of T cells and subsequently T-cell cytotoxicity when autologous glioma cells were introduced [81, 82]. Furthermore, a robust cytotoxic (CTL) and memory T-cell lymphocyte infiltration into intracranial tumors was observed in murine models of glioma following vaccination and peripheral infusion of tumor antigen-primed DCs, favoring a Th1 lymphocyte activation state, capable of homing to and expanding within tumor tissue [83].
Though many investigative protocols for DC-based vaccination of malignant glioma differ with regard to protocol specifics, most involve extraction of DC precursors in the form of peripheral blood mononuclear cells (PBMCs); exposure to tumor-associated formulation in the presence of GM-CSF and IL-4, both known to direct APC maturation; and reintroduction though subcutaneous, intradermal, intranodal, or intratumoral injection.
The mechanism by which tumor-associated antigens are loaded in vitro into DCs is of critical importance. Multiple DC-loading strategies have been employed, including the use of autologous tumor lysates, formulations of apoptotic material following ATC irradiation, and purified or synthetic tumor-associated peptide antigens [84–86].
A potential advantage of loading strategies which do not isolate individual antigens, such as the use of autologous tumor lysates, is the induction of an immune response against multiple tumor epitopes, though likely at the expense of non-tumor-specific cross-reactivity and subsequent autoimmune toxicity. Those strategies using distinct and tumor-specific antigens, either purified or synthetic, limit the activation of cross-reactive lymphocytes, allowing for the escape of non-expressing clonal populations.
To date, 15 clinical trials including 316 total patients have evaluated the use of DC-based vaccination in the treatment of malignant gliomas including primary and recurrent GBM, anaplastic astrocytoma (AA), anaplastic oligoastrocytoma (AOA), and anaplastic oligodendroglioma (AO) (reviewed in [70]): eight phase I trials, six phase I/II trials, and one phase II trial. Table 18.1 summarizes the vaccination details and clinical results of these trials. Across all included in these trials, only one patient suffered grade IV neurotoxicity resulting from a large residual tumor and perilesional edema [90], highlighting the safety and feasibility of antigen-primed DC vaccinations for CNS cancers. Immune response was largely evaluated by delayed-type hypersensitivity (DTH); increased proportions of CTLs, NKs, and memory T cells both in peripheral blood and as infiltrating lymphocytes in subsequent tumor resections; increased tumor cell reactivity of postvaccination extracted PBMCs exposed to ATC in vitro; and increased presence of IFN-γ both peripherally and within the tumor-infiltrating lymphocytes. Over half of the patients enrolled in these trials demonstrated some evidence of an immune response following vaccination, and all 15 studies reported a survival benefit following vaccination (Table 18.1). Moreover, two of these trials focusing on patients with GBM demonstrated an improved response to chemotherapy delivered in a second phase following DC-based vaccination, suggesting an exciting potential for synergy with these treatments [85, 96].
Table 18.1
List of clinical trials utilizing dendritic cell vaccinations in patients with malignant gliomas
Patients | Phase | Route | Antigen format | Immune response | Clinical response | References |
|---|---|---|---|---|---|---|
22 patients (13 recurrent GBM, 5 AA, 3 AO, 1 AOA | Phase I/II | Intranodal + intramuscular injections of poly-ICLC | Synthetic peptides for GAAs | Induced positive immune responses against at least one of the GAAs in PBMCs in 58 % of patients (after 4 vaccinations). Significant upregulation: interferon-alpha and CXCL10 | 4 recurrent GBMs are progression-free for at least 12 months; 1 CR (recurrent GBM). Median TTP, 4 months | Okada (2011) |
23 patients (15 newly diagnosed GBM, 8 recurrent GBM) | Phase I | Intradermal + intramuscular injections of poly-ICLC | Autologous tumor lysate + imiquimod or poly-ICLC | No dose-limiting toxicity. Tumor samples with a mesenchymal gene expression signature had a higher number of CD3+ and CD8+ tumor-infiltrating lymphocytes | Newly diagnosed: median OS, 35.9 months, with a mean follow-up time of more than 4 years and 1-, 2-, and 3-year survival rates of 93, 77, and 58 %, respectively. Recurrent: median OS was 17.9 months from the time of initial glioblastoma diagnosis. OS was significantly longer for those who received DC vaccination at initial diagnosis compared with those who enrolled in this trial at the time or recurrence | Prins (2011) |
8 patients (newly diagnosed GBM) | Phase I/II | Intradermal | Autologous tumor lysate | DTH (2/5) increased CD8+/CD25+ in PBL (6/7) ATR PBMC (5/8) (IFN-gamma ELISPOT) | Median OS, 24 months | Ardon (2010) |
45 children (23 recurrent GBM, 5 AA, 1 AOA, 16 other HGG) | Phase I | Intradermal | Autologous tumor lysate + imiquimod | No data available | Median PFS for relapsed GBM, 4.3 months; median OS for relapsed GBM, 12.2 months | Ardon (2010) |
12 patients (newly diagnosed GBM) | Phase I | Intradermal | EGFRvIII antigen + KLH | DTH EGFRvIII (5/9); DTH KLH (9/9); ATR PBMC (10/12) (EGFRvIII-induced proliferation) | Median OS, 22.8 months | Sampson (2009) |
56 patients (recurrent GBM) | Phase I/II | Intradermal | Autologous tumor lysate | DTH (9/21 at time of diagnosis, 9/17 after 2 vaccinations) | 3-month PFS; OS, 9 months; 24-month OS, 14.8 %; total resection is a predictor for better PFS; younger age and total resection are predictors for better OS | De Vleeschouwer (2008) |
34 patients (23 recurrent GBM, 11 newly diagnosed GBM) | Phase II | Subcutaneous | Autologous tumor lysate | Postvaccine antigen-directed IFN-gamma response in PBMCs (17/34); DTH test resulted in cutaneous GBM in 1 patient (DTH was subsequently discontinued) | Newly diagnosed: 8/17 (47 %) vaccine responders versus 3/15 (20 %) nonresponders. Recurrent: TTS, 621 ± 81 and 402 ± 49 days; TTP, 28 ±94 and 142 ± 22 days (8 responders and 13 nonresponders); TTP, 343 ± 116 and 136 ± 19 days (8 responders and 15 nonresponders) | Wheeler (2008) |
24 patients (18 recurrent GBM, 6 grade III glioma) | Phase I/II | Intradermal or intradermal + intratumoral (Ommaya reservoir) | Autologous tumor lysate | DTH to tumor lysate (8/24); ATR PBMC (7/24) (IFN-gamma ELISPOT) | 1 PR, 3 MR, 6 SD (GBM); 4 SD (grade III glioma); median OS, 16 months versus 13.3 months; longer survival if DC maturation or IC injection. One grade IV neurotoxicity event (stupor) observed | Yamanaka (2005) |
12 patients (7 newly diagnosed GBM, 5 recurrent GBM) | Phase I | Intradermal | Acid-eluted tumor-associated peptides | CTL response (6/12); tumor infiltration CD8+ CD45RO+ cells (4/8) | Median TTP, 19.9 months, OS 18 to >58 months; median OS, 25.8 months. 1PR; median OS, 23.4 versus 18.3 months. No dose-limiting toxicity observed | Liau (2005) |
14 patients (1 newly diagnosed GBM, 9 recurrent GBM, 4 AA) | Phase I | Subcutaneous | Autologous tumor lysate | Increased IFN-gamma RNA in PBMC (6.10) ATR T cells (4/9) (HER-2, GP100, MAGE-1 tetramers); CD8+, CD45RO+ cell infiltration (3.6) | Median survival, 33.3 versus 7.5 months (8/9 recurrent GBM) | Yu (2004) |
15 patients (6 recurrent GBM, 7 AA, 2 AOA) | Phase I | Intradermal | DC fusion with autologous glioma cells | DTH to tumor lysate (15/15); increased cytotoxic activity (2/15); increased intracellular IFN-gamma in CD8+ T cells (1/15) | 1 SD (GBM); 3 PR, 1 MR (AA); 1 PR, 1 SD (AOA) | Kikuchi (2004) |
7 patients (2 recurrent GBM, 1 AA, 4 other HGG) | Phase I | Intradermal | Autologous tumor RNA | No antitumor responses (0/3) (IFN-gamma ELISA) | 1 PR (1XA); 4 SD (1AA, 3 other HGG) | Caruso (2004) |
25 patients (newly diagnosed GBM: 13 plus chemotherapy, 12 without chemotherapy) | Phase I/II | Intradermal | Autologous tumor lysates or peptide elutions | Vaccine alone: ATR PBMC (4/11) vaccine + chemotherapy: ATR PBMC (4/13) (lytic activity and IFN-gamma PCR) | Vaccine or chemotherapy alone: 24-month survival 8 %; vaccine + chemotherapy, 3 PR; 24-month survival 42 % | Wheeler (2004) |
10 patients (7 recurrent GBM after radiotherapy, 3 recurrent grade III glioma) | Phase I/II | Intradermal and/or intratumoral (Ommaya) | Autologous tumor lysate | Increase in NK cells in PBMCs (5/10); DTH to tumor lysate (3/10); increased T-cell-mediated antitumor activity (2/10) | 2 MR, 2SD (GBM), 2SD (grade III glioma); OS >50 months | Yamanaka (2003) |
9 patients (7 newly diagnosed GBM, 2 AA after radiotherapy) | Phase I | Subcutaneous | Tumor-specific MHC I-associated peptides | Systemic CTL cytotoxicity against tumor (4/9) (lytic activity); tumor infiltration: CD4+, CD8+, CD45RO+ cells (2/4) | Prolonged median survival compared to control group: 15.2 versus 8.6 months (GBM) | Yu (2001) |
Despite the variability across these trials, salient insights include the safety of DC-based approaches to CNS tumor vaccination and the feasibility of these immune strategies as some features of an elicited immune response were demonstrated in over half of all patients enrolled. Other interesting results include the improved success of matured DC vaccinations generated by combining antigen priming with maturation factors such as TNF-α, Toll-like receptor (TLR) ligands, or IFN-γ and the potentially synergistic effect of DC-based vaccination and chemotherapy in treating brain tumors. Details regarding precise protocols for loading of DCs, amount and site of injection, and composition of accompanying adjuvants remain to be optimized.
An additional trial evaluated the use of DC-based immunotherapy in 45 pediatric patients with high-grade glioma, medulloblastoma, primitive neuroectodermal tumors (PNETs), ependymoma, and atypical teratoid-rhabdoid tumors (ATRTs) [97]. The authors utilized autologous tumor lysates to load PBMC-derived DCs and delivered these by intradermal injections followed by two subsequent boost vaccinations of tumor lysate. No severe adverse effects occurred in those patients with high-grade gliomas and ATRTs, and additionally, overall survival was increased compared to historical controls in those two tumor types. In those patients with PNET and medulloblastoma tumors, vaccinations were discontinued due to adverse effects. These findings show the potential of the DC-based immunotherapy to pediatric brain tumors, but data regarding efficacy remains preliminary and poorly controlled.
The optimization of tumor-associated antigen-loading strategies is under active exploration. A recent study compared specific antigenic peptide-loaded vs. autologous tumor lysate-loaded DC vaccines for treating malignant glioma [98].
Twenty-eight patients were treated with autologous tumor lysate-pulsed DC vaccines, whereas six patients were treated with glioma-associated antigen peptide-pulsed DCs, utilizing a synthetic formulation of four epitopes known to be expressed on malignant gliomas. These antigens included survivin, HER-2/neu, gp100, and TRP-2, which are present in approximately 60, 80, 60, and 50 % of malignant gliomas specimens, respectively [27]. No adverse events were reported in either study group. The median survival of patients on the autologous tumor lysate-DC trial was 34.4 months, whereas that of patients on the synthetic glioma-associated antigen-DC group was significantly different with a median survival of 14.5 months [27]. Though limited to small cohorts under individual protocols, these results support the use of autologous tumor lysate preparation in priming DCs for vaccination in CNS cancer. The authors also noted a significant correlation between decreased Treg ratios (pre- vs. postvaccination) and overall survival, evident in both study groups.
18.8.2.3 Antigen-Specific Peptide Strategies
In contrast to the DC-based techniques discussed above, direct peptide vaccines rely on the ability of host APCs in the periphery to process, migrate, and present the introduced antigens. Extensive preclinical analysis has demonstrated the ability of peripheral APCs to activate T cells within lymph nodes regional to the site of injection in animal models of brain tumors [70, 99]. Refinements to direct antigen vaccination strategies have demonstrated the utility of adjuvant compounds such as keyhole limpet hemocyanin (KHL) as an immunogenic peptide carrier protein [100] and GM-CSF as a mitogenic stimulus for APCs [101], both of which ultimately augment antigen presentation.
The selection of tumor-associated peptides to enable selective tumor cell targeting with minimal secondary autoimmunity is critical to the success of any vaccination utilizing target peptide sequences, both in direct peptide injection and in specific antigen-primed DC infusion. Considerable effort has been expended in identifying antigens differentially or exclusively expressed in CNS tumors, including genes only normally expressed during embryological development, differently spliced or mutated genes, and genes giving rise to fusion proteins, which result from the general genetic instability of transformed cells, as well as housekeeping or metabolic pathway antigens which may be exclusive to tumor cells [90]. Nevertheless, an increasing appreciation of intratumor clonal heterogeneity [102] complicates effective targeting of a clinically significant proportion of tumor cells through a specific antigen vaccination strategy.
The National Cancer Institute (NCI) recently performed an in-depth review of 75 general tumor-associated antigens to evaluate their potential as targets for immunotherapy [103]. The potential of tumor antigens to serve as targets for immunotherapy was graded according to the following criteria: therapeutic function, immunogenicity, oncogenic function, specificity, expression level in tumors, in cancer stem cells, percentage of tumors that express it and cellular localization of the protein. Based on this, the antigens that showed the most potential for immunotherapy where. The highest-ranked antigens included WT-1, MUC1, LMP2, HPV E6/E7, HER2/neu, EGFRvIII, melanoma antigen-encoding (MAGE)-A3, and NY-ESO-1. While expression of some of these antigens in CNS cancer is well established, such as the expression of EGFRvIII in GBM [104], the presence or absence of others in CNS cancers warrants future investigation. Additional insight into CNS cancer-specific antigen targets for immune-based therapy has come from tumor antigen investigations in melanoma [105]. The genes MAGE-1 [106], MAGE-E1 [107], MAGE-3, and glycoprotein-240 (a cell surface glycoprotein of 240,000 molecular weight present in most melanomas) [108] were expressed in many different glioma subtypes but never in normal brain tissue and therefore present as potential targets for CNS tumor-specific immunotherapy. Many additional CNS cancer-associated antigens have been described as potential tumor-selective targets for immunotherapy in a variety of CNS tumor types; these include but are not limited to tenascin, homo sapiens testis (HOM-TES)-14 (also known as stromal cell-derived protein (SCP)-1), HOM-TES-85, synovial sarcoma X chromosome breakpoint (SSX)-1, SSX-2, GAGE-1, SRY-related high-mobility group (HMG)-box-containing gene (SOX)-5, cancer testis antigen 6, IL-13 receptor a2, ephrin (Eph) A2, antigen isolated from immunoselected melanoma (AIM)-2, squamous cell carcinoma antigen recognized by T cells (SART)1, SART3, and kinesin superfamily protein (KIF)1C and KIF3C [70]. See Table 18.2 for a list of GBM-associated antigens under preclinical or clinical investigation in tumor vaccines. Ongoing effort to characterize these many relevant antigens in various CNS cancer subtypes will hopefully yield firm footing of which to launch future antigen-specific immune-based therapies.
Table 18.2
List of glioma-associated antigens (GAAs) which may serve as immunotherapeutic targets
GAA | Characteristic/potential function | References |
|---|---|---|
AIM2: absent in melanoma 2 | AIM2 could be used as a tumor antigen target for monitoring vaccine trials or for developing antigen-specific active immunotherapy for glioma patients | Okada (2009), Liu (2004) |
BMI1: BMI1 polycomb ring finger oncogene | Expressed in human GBM tumors and highly enriched in CD133+ GSC cells | Abdough (2009) |
COX-2: cyclooxygenase-2 | Overexpressed in many tumors including CD133+ GSC cells; COX-2 inhibitor celecoxib will become a nice weapon for GBM therapy | Shono (2001) |
TRP-2: tyrosinase-related protein 2 | Highly expressed in GSCs | Saikali (2007), Driggers (2010) |
GP100: glycoprotein 100 | Melanocyte lineage-specific antigen, expressed in GSCs as well | Saikali (2007), Driggers (2010) |
EGFRvIII: epidermal growth factor receptor variant III | EGFRvIII is the most prevalent of several EGFR mutations found in human gliomas and is expressed in 20–25 % of GBM. GSC-associated antigen | Saikali (2007), Driggers (2010) |
EZH2: enhancer of zeste homologue 2 | Upregulated in malignant gliomas in GSC cells | Orzan (2011) |
LICAM: human L1 cell adhesion molecule | Highly expressed in GSCs. Invasion-associated proteins | Cheng (2011) |
Livin and Livin β | Livin β was more related with the high survival rate. It is a cancer-associated member of the inhibitor of apoptosis protein (IAP) | Jin (2010) |
MRP-3: multidrug-resistance protein 3 | GBMs overexpress MRP-3 at both mRNA and protein levels. Multidrug-resistance protein 3 has potential correlation with survival. Highly expressed in GSC cells as well | Driggers (2010), Kuan (2010) |
Nestin | Nestin plays important roles in cell growth, migration, invasion, and adhesion to extracellular matrices in glioma cells. Overexpressed in GSCs | Ishiwata (2011) |
OLIG2: oligodendrocyte transcription factor 2 | GSC marker, OLIG2 is highly expressed in all diffuse gliomas. Immunohistochemistry and microarray analyses demonstrated higher OLIG2 in anaplastic oligodendrogliomas versus glioblastomas, which are heterogeneous with respect to OLIG2 levels | Ligon (2004) |
SOX2: SRY-related HMG-box 2 | SOX2 expression and amplification in gliomas and GSC cell lines | Xu (2009)
Related posts: Immunopathology and Immunotherapy of Lymphoblastic Leukaemia
Immunotherapy for Pediatric Solid Tumors
Immunopathology of Specific Cancers in Males and Females and Immunotherapy of Prostate and Cervical Cancer
Immunology and Immunotherapy of Ovarian Cancer
Immunotherapy of Renal and Bladder Cancers
Immunology and Immunotherapy of Breast Cancer Immunopathology and Immunotherapy of Lymphoblastic Leukaemia
Immunotherapy for Pediatric Solid Tumors
Immunopathology of Specific Cancers in Males and Females and Immunotherapy of Prostate and Cervical Cancer
Immunology and Immunotherapy of Ovarian Cancer
Immunotherapy of Renal and Bladder Cancers
Immunology and Immunotherapy of Breast Cancer
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|