The observation that lymphocytes from two unrelated individuals can stimulate each other to blast formation when cultured together (mixed lymphocyte culture (MLC) assay), and that the antigens responsible for this stimulation are inherited together with HLA antigens, led to the discovery of HLA-D antigens (Bach and Hirschhorn 1964; Bain et al. 1964), which were later detected serologically on B cells and named DR antigens (van Rood et al. 1975, 1976; van Rood and van Leeuwen 1976). HLA-DR molecules together with HLA-DQ and HLA-DP constitute the classical class II molecules.
The ability to study HLA genes and their alleles at the molecular level has advanced the knowledge of the HLA genes enormously. It is now known that the HLA genes constitute a coding sequence in a region that spans about 4000 kilobases. The genes and pseudogenes are grouped in three subregions of the short arm of chromosome 6 (Horton et al. 2004). Not only is the region now known ‘nucleotide by nucleotide’ at the genome level, but also hundreds of alleles of the different loci have been sequenced in the population. Most HLA typing is now done at the DNA level.
Human Leucocyte Antigen: the Human Major Histocompatibility Complex
The name MHC refers to the ability of the genes of this genomic region to determine graft rejection between individuals of the same species. In the 1960s, the HLA genes, first discovered through leucocyte agglutination, were established as the genes responsible for graft rejection in man. The physiologic function of these molecules was determined during the following decade: presentation of antigens to T cells (Zinkernagel and Doherty 1974). The mechanism of this ‘HLA restriction’ was first explained by Townsend and co-workers (1986), who showed that synthetic peptides could be presented to the T cell. The final explanation of how this peptide could be presented to the T cell awaited the discovery of the structure of the HLA molecules in 1987 and the crystallization of a T-cell receptor bound to an MHC molecule.
The extremely polymorphic, closely linked genes of the HLA system are located in a region that spans about 4000 kilobases (kb) on the short arm of chromosome 6 (Breuning et al. 1977). Moving from centromere to telomere, the class II genes are separated from the class I genes by a number of functionally unrelated genes (and pseudogenes) that encode the complement factors C2, C4a and C4b heat shock proteins, cytokines and enzymes (class III genes) (Figure 13.1).
The MHC is the most diverse region in the human genome. After three decades of maps of ever increasing elaboration, the complete sequence of the human MHC was published in 1999 by the MHC Sequencing Consortium (1999). An integrated map of the extended MHC has is also available (Horton et al. 2004).
Class I Region
HLA-A, -B and -C code for the heavy chain of the MHC class I molecules expressed on most cells. HLA-F, -G and -E code for the heavy chain of non-classical class I molecules, with highly specialized functions. The MIC genes or human MHC class I chain-related genes encode stress-inducible proteins implicated in the regulation of NK cell activity. HFE is a class I-like gene located approximately 4 Mb telomeric of HLA-F and responsible for most hereditary haemochromatosis.
Class III Region
TNFB and -A code for tumour necrosis factors, HSP genes for heat shock proteins, C2, Bf and C genes for proteins of the complement system, and P450-C21B for a steroid 21-hydroxylase.
Class II Region
The HLA-DRBA1, -DQA1 and -DPA1 genes code for alpha chains of the DR, DQ and DP class II molecules. HLA-DRB1, -DQB1 and -DPB1 code for the beta chain of the DR, DQ and DP class II molecules. In addition to HLA-DRB1, which codes for the primary HLA specificities such as DR1, DR2, DR4, etc., other DRB genes code for the beta chain of the specificities DR52 (DRB3), DR53 (DRB4) and DR51 (DRB5) not present in all haplotypes. The DO and DM molecules regulate the loading of exogenous peptides into class II molecules. LMP2 and LMP7, which encode the subunits of the proteasome, and TAP1 and TAP2, which encode a peptide transporter, are involved in the processing and presentation of antigens by class I molecules.
HLA Class I and II Molecules: Structure and Function
HLA molecules engage two distinct arms of the T-cell-mediated immune response. MHC class I molecules present antigen to cytotoxic T cells (CTLs), whereas MHC class II present to helper T cells. Antigens are not presented by HLA molecules as intact proteins. The antigen must first be degraded to peptide fragments and presented in the context of the HLA molecule to the T-cell receptor.
HLA Class I Molecules
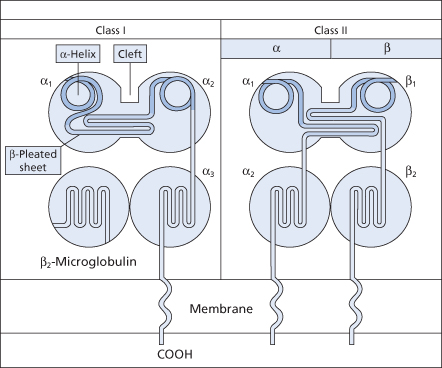
HLA-A, -B and -C genes produce a transmembrane glycosylated polypeptide of molecular weight 4300 (the α or heavy chain) linked non-covalently to β2-microglobulin, a non-polymorphic and nonglycosylated polypeptide of molecular weight 1200 (the β or light chain), which is encoded by a gene on chromosome 15 (Snary et al. 1977a; Barnstable et al. 1978). The extracellular part of the heavy chain consists of three domains: α1, α2 and α3 (Figure 13.2).
Figure 13.2 Structure of class I and class II HLA molecules showing domains and transmembrane segments.
(Source: Roitt and Delves 2001. Reproduced with permission of John Wiley & Sons.)
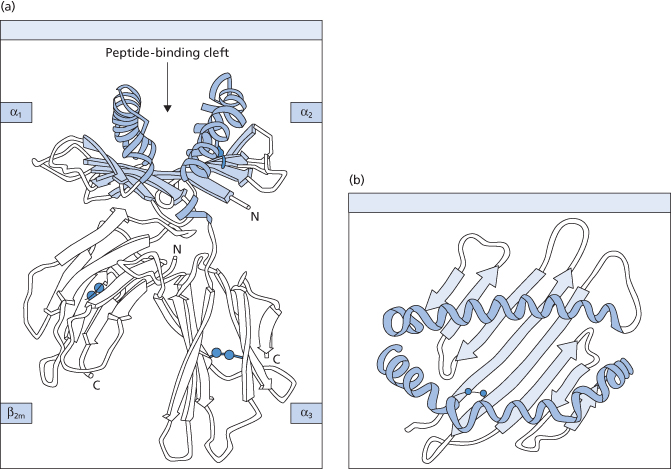
The three-dimensional structure of class I molecules has been revealed by X-ray crystallographic analysis, first of HLA-A2 (Figure 13.3) and subsequently of HLA-A68 and HLA-B27 (Bjorkman et al. 1987; Garretti et al. 1989; Madden et al. 1991). The α3 and β2m domains have tertiary structures similar to domains in the constant region of the immunoglobulins. The top of the molecule is formed by pairing the α1 and α3 domains, which together form the antigen peptide-binding groove. The majority of the polymorphic determinants in class I molecules occur on the floor and α helices of this cleft (Bjorkman et al. 1987) (see Figure 13.3). The class I molecules specifically bind peptides of defined length, usually 6–10 residues (Falk et al. 1991). All peptides bind similarly with their N- and C-termini sequestered in the binding groove by a network of hydrogen bonds to residues conserved in all class I glycoproteins (Madden and Wiley 1992). In addition, there are allele-specific binding pockets with a strong preference for a few side-chains at some positions of the peptide. This explains the correlation between class I polymorphism and the affinity of peptide binding (Falk et al. 1991). Generally, these peptides derive from self-proteins, but in virus-infected cells the peptides from the pathogen may be processed in the cytosol and migrate with the HLA molecules to the cell surface (Pamer and Cresswell 1998). Tumour antigens can be detected in the same way. Class I molecules are expressed on most cells and they inform the scanning CTL about the status of potential target cells for destruction. Peptide epitopes presented by them can only be recognized by CTL if these (1) have a specific receptor for the antigen and (2) the same HLA class antigens as the target cell. This phenomenon, known as HLA restriction, was first described in the mouse (Zinkernagel and Doherty 1974). The HLA-A, -B and -C antigens are expressed on all nucleated cells except spermatozoa and placental trophoblast. The antigens are also found on platelets and some class I antigens have been detected on red cells. The number of class I molecules on various cells differs and, particularly on platelets, some of the antigens are weakly expressed.
Figure 13.3 Schematic representations of the crystallized structure of the HLA-A2 molecule. (a) The four domains, with the α1 and α2 domains forming a putative peptide-binding region. (b) Top surface of the molecule. The putative antigen-binding groove is shown, made up of a β-pleated sheet flanked by two α-helices.
(Source: Bjorkman et al. 1987. Reproduced with permission of Nature.)
HLA Class II Molecules
All typical class II molecules consist of two transmembrane glycoprotein chains of molecular weight 33 kDa (the heavy or α-chain) and 28 kDa (the light or β-chain) respectively (Snary et al. 1977b). The extracellular component of both chains consists of two distinct domains: α1, α2 and β1, β2. The domains distal to the cell surface carry most of the polymorphic determinants. The constant domain near the cell surface is very similar to the constant domain of the immunoglobulin heavy chains (Shackelford et al. 1982; see also Figure 13.2).
The crystal structure of class II molecules (DR1) is similar to that of class I molecules; polymorphic determinants of class II molecules are also clustered in the antigen peptide-binding groove (Brown et al. 1993). In contrast with class I molecules, class II molecules bind longer peptides with no apparent restriction on peptide length (Rudensky et al. 1991). The peptides bind to the groove as a straight extended chain with a single pronounced twist. Hydrogen bonds along the main chain of all peptides interact with residues from the α-helical regions and the β-sheet in the peptide-binding groove and thus provide a binding component that is independent of the sequence of the peptide. Twelve hydrogen bonds on the peptide bind to determinants encoded by residues conserved in most class II alleles and this suggests that peptides bind to class II molecules by a universal mode. However, particular side-chains of the peptide are accommodated in polymorphic pockets in the binding groove that determine specific binding of peptides and thus the affinity of the peptide class II molecule bond (Stern et al. 1994). The expression of class II antigen is restricted to B cells and to antigen-presenting cells such as macrophages, dendritic cells and Langerhans cells. Class II antigens are also present on activated T lymphocytes and some tumour cells (Winchester and Kunkel 1979).
The cells that express class II molecules, specialized antigen-presenting cells (APCs), such as dendritic cells, mononuclear phagocytes and B cells, bind exogenously derived peptides of 9–22 residues. In the case of macrophages and B cells, the HLA molecule–antigen complex is assembled within intracellular organelles. With all APCs the antigen peptides are held in a groove in HLA class II molecules, and this plasma membrane-bound compound antigen is recognized by helper T lymphocytes via their T-cell receptors during immunosurveillance. The polymorphic HLA determinants in the peptide-binding groove of the class II molecule strongly influence peptide binding.
Dendritic cells, which express HLA class II antigens particularly well, are the APCs that present antigen to helper T cells to induce a primary immune response. Memory T cells can be stimulated by macrophages, B cells and even by free antigens (Berg et al. 1994). Class II antigen complexes instruct the helper T-cell system to initiate the humoral immune response and assist in the cellular immune process; for this reason, class II genes are often referred to as ‘immune response genes’.
HLA Genes and Antigens
The linkage between the HLA genes is so strong that crossing over between them is rare; therefore the alleles of the HLA genes present on one chromosome usually segregate together within a family. The two alleles of each individual gene are expressed co-dominantly (e.g. HLA-A1, -A11). The set of HLA alleles present on a single chromosome is known as a haplotype. Siblings who inherit the same haplotypes from their parents are thus HLA identical, unless crossing over between HLA genes has occurred.
As crossing over within the HLA region is rare, with random assortment equilibrium should be reached in a population over a long period of time; particular combinations of alleles at, for example, the A and B loci or at the loci of the D region should not be more common than predicted from the product of their relative frequencies in the population. However, in any given population, certain combinations of alleles or haplotypes are more frequent than expected, a phenomenon known as ‘linkage disequilibrium’. For example, the frequency of HLA-A1-B8 in European white people is 8.8%, whereas the expected frequency of this haplotype, based on the individual frequencies of A1 and B8, is 1.6%. Selective pressures that affect survival or reproductive capacity usually drive linkage disequilibrium. Patterns of linkage disequilibrium vary in different populations.
Nomenclature
A history of the development of HLA system nomenclature has been compiled by Bodmer (1997). The IMGT/HLA Sequence Database continues to act as the official repository for HLA sequences named by the WHO Nomenclature Committee for Factors of the HLA System. The database contains sequences for all HLA alleles officially recognized by WHO and provides users with online tools for their retrieval and analysis. New releases of the database are now made monthly. The database may be accessed via the worldwide web at www.ebi.ac.uk/imgt/hla.
For the non-aficionado, nomenclature remains a challenge, because two systems remain in general use. The older serological nomenclature relies on identification of antigens on the leucocyte surface (HLA antigens) (Tiercy et al. 2002). The following terms are used for the ‘classical’ HLA antigens: for class I, the capital letters –A, –B –C are appended to identify the locus; for class II, the prefix D followed by a letter (-DR, -DQ, -DP) for the subregion and by the letters A or B to indicate whether the gene codes for the α- or β-chain, for example DRA, DQB, etc. The letters are followed by a number that identifies epitopes determined by alloantibodies or less often alloreactive cytotoxic T cells.
New sequence-based nomenclature was introduced in 2010 (Marsh et al. 2010). Sequence-based nomenclature separates the HLA locus with an asterisk (*) followed by up to four sets of digits separated by colons (Figure 13.4).
Figure 13.4 HLA nomenclature. The first two digits describe serologically defined antigens; the next two or three digits list the subtypes. The third set of digits designate alleles that differ only by synonymous nucleotide substitutions, ‘silent’ or non-coding substitutions. The fourth set of digits complete the number of the allele as defined by molecular techniques that differ only by sequence polymorphisms in the non-coding region. Additional suffixes may be added to an allele to indicate its expression status: ‘N’ (Null), ‘L’ (low), ‘S’ (soluble) ‘C’ for an allele product in the cytoplasm but not on the cell surface. Image used courtesy of Professor SGE Marsh, HLA Informatics Group, Anthony Nolan Research Institute, London, UK, http://hla.alleles.org/nomenclature/naming.html
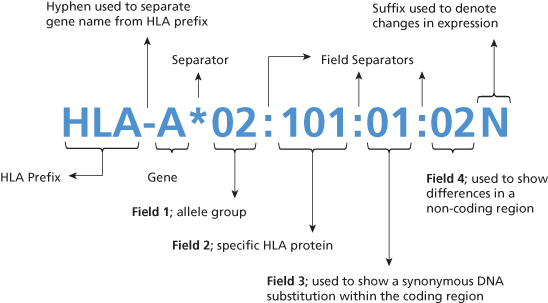
The first two digits describe the serologically defined antigen with which the allele is (or alleles are) most closely associated; the next two or three digits list the subtypes in the order in which DNA sequences have been determined. Alleles whose numbers differ in the two sets of digits must differ in one or more nucleotide substitutions that change the amino acid sequence of the encoded protein. The third set of digits designate alleles that differ only by synonymous nucleotide substitutions, ‘silent’ or non-coding substitutions. The fourth set of digits complete the number of the allele as defined by molecular techniques that differ only by sequence polymorphisms in the non-coding region. W (‘workshop’) was used previously to indicate that the specificity was provisional, but now all serological specificities will be named on the basis of correlation with an identified sequence. The letter ‘w’ can therefore be dropped with three sets of exceptions: (1) Bw4 and Bw6 to distinguish them as epitopes from those encoded by other alleles of the HLA-B gene; (2) the C antigens for which the w is retained throughout to avoid confusion with the nomenclature of the complement system; and (3) the Dw specificities defined by the MLC assay; and the DP specificities defined by a secondary response of T lymphocytes that had been primed by a first step in the MLC (primed lymphocyte typing) (Bodmer et al. 1992). In addition to the unique allele number, additional suffixes may be added to an allele to indicate its expression status: ‘N’ (Null) for alleles that are not expressed, ‘L’ for low cell surface expression, ‘S’ for an allele specifying a protein which is expressed as a soluble molecule that is secreted but is not present on the cell surface, ‘C’ for an allele product in the cytoplasm but not on the cell surface.
One of the non-classical HLA class I genes, the HLA-G gene, encodes a non-polymorphic α-chain with a shortened cytoplasmic segment. The HLA-G molecule is expressed only on the trophoblast, which suggests that it may have a role in embryonic development or fetal–maternal immune interactions, or both (Geraghty et al. 1987; Kovats et al. 1991). Class I antigens are detected in a lymphocytotoxicity test, using either alloantibodies or human or murine monoclonal antibodies.
Class II Genes and Antigens
The polymorphism of the class II genes is much greater than detected on their products by serological typing and by the MLC assay. Studies at the DNA level have shown that, in addition to the classical DR, DQ and DP series of genes, there are several other non-classical class II genes in the D region: DOA, DOB, DNA and the DMA and DMB genes. In addition, in the class II chromosomal region, there are four genes, TAP1 and TAP2, and LMP2 and LMP7, which encode molecules involved in antigen processing (see below).
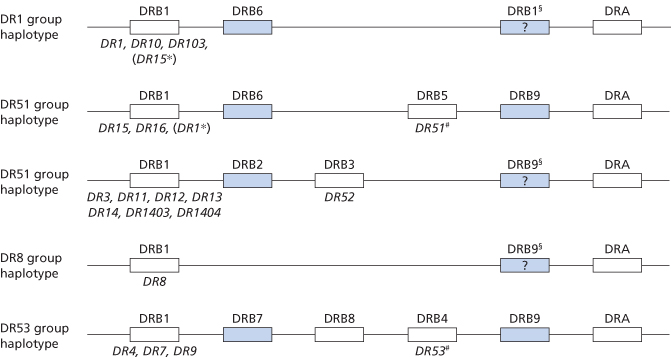
In the DR subregion there is a single α-chain gene (DRA) with two alleles that, however, do not encode a polymorphism on the α-chain. There are nine DRB genes, five of which are pseudogenes (DRB2, DRB6, DRB7, DRB8 and DRB9). Seven of the DRB genes (except DRB1 and DRB9) are restricted to certain DR haplotypes. The genomic organization of the DR region is shown in Figure 13.5. The DRB1, DRB3, DRB4 and DRB5 genes encode four separate β-chains. DRB1 encodes the major DR antigens, whereas the DRB3 gene codes for DR52, DRB4G or DR53 and the DRB5 gene for DR51.
Figure 13.5 Genomic organization of the HLA-DR region and encoded products (specificities). Pseudogenes are indicated by shaded boxes, expressed genes by open boxes. The serological specificity encoded by a gene is shown underneath in italics. *, rarely observed haplotypes; #, DR51 and DR53 may not be expressed on certain haplotypes; §, the presence of DRB9 in these haplotypes needs confirmation.
(Source: Bodmer et al. 1992. Reproduced with permission of John Wiley & Sons Ltd.)
In the DQ subregion there are two α genes: DQA1, which encodes an α-chain and DQA2, which is not known to be expressed. There are three B genes: DQB1, which encodes a β-chain and DQB2 and DQB3, not known to be expressed. DQA1 and DQB1 are polymorphic but only the products of the DQB1 alleles have been serologically recognized (DQ antigens).
In the DP subregion there are two α and two β genes: DPA1, DPA2, DPB1 and DPB2. DPA1 and DPB1 encode an α- and a β-chain respectively, whereas DPA2 and DPB2 are pseudogenes.
Additional HLA Class II Genes
The genes DOB and DNA located between the DP and DQ subregions encode a β- and α-chain, respectively, whose function is as yet unknown. The genes DMA and DMB encode an α- and a β-chain, which associate to form a class II molecule that contains a peptide-binding groove involved in antigen presentation (Kelly et al. 1991). The DM genes are polymorphic, but the polymorphism is limited and the resulting antigenic determinants occur only on the area of the extracellular portion of the protein that is proximal to the cell membrane. They therefore do not occur in the peptide-binding groove and thus do not affect antigen presentation, in which the DM molecule probably has a specialized function (Sanderson et al. 1994).
Non-HLA Genes Involved in Antigen Processing
The TAP1 and TAP2 genes, located between DBO and DNA, do not encode typical class II proteins but instead encode an important peptide transporter molecule involved in the endogenous processing of antigen (Spies et al. 1991). The TAP genes are polymorphic (Colonna et al. 1992; Powis et al. 1992a,b, 1993). This polymorphism may affect antigen processing and thus the immune response, but at present this is speculation. The LMP2 and LMP7 genes, located near the TAP genes, encode proteasomes that affect the degradation of antigen molecules to peptides (Cerundolo et al. 1995).
The class II antigens are detected by alloantibodies and, in some cases, also by monoclonal antibodies, using a complement-dependent cytotoxicity test on isolated B lymphocytes or a two-colour immunofluorescence test on unseparated cells (van Rood et al. 1976).
As mentioned above, a polymorphism (Dw) encoded by the D region has been defined by using homozygous typing cells in the mixed lymphocyte culture (MLC). The exact relationship between Dw determinants and the polymorphic determinants encoded by the DR, DP genes is not known. However, a strong correlation has been observed between matching and mismatching for DR polymorphic DNA sequences and reactivity in the MLC. The lymphocytes of all DR mismatched pairs were reactive and 37% of the matched pairs were non-reactive in the MLC. The MLC reactivity of 63% of the matched pairs may be due to unrecognized DR alleles (Baxter-Lowe et al. 1992), but incompatibility for HLA-DP specificities has also been found to induce significant proliferation in the primary MLC in HLA-A, -B, -DR and -DQ identical subjects (Olerup et al. 1990). HLA-DQA1 or –DQB1 allele differences are not important in the primary MLC among otherwise HLA identical, unrelated subjects (Termijtelen et al. 1991).
For a list of the very numerous class II alleles, antigens and determinants agreed by the WHO Nomenclature Committee, see Marsh (2003).
Other Genes in the HLA Region
As the products of these genes are not leucocyte antigens and are not involved in antigen presentation by HLA molecules, they will not be discussed further.
Crossreactions in the HLA System
Sera from subjects alloimmunized against HLA antigens are frequently crossreactive, as shown by the following example: a given serum may react with two different serologically defined antigens, for example HLA-B51 and HLA-B52, but antibodies recognizing these two specificities individually cannot be separated from the serum. The antibodies in such a serum are in fact directed against a different antigen, HLA-B5 in this example, which occurs together with B51 and B52. Thus the antibodies (anti-B5) crossreact with B51- and B52-positive cells.
This kind of crossreactivity is explained by the multiple mutations within the HLA genes. A single allele of a gene may code for different, separate polymorphisms on the single HLA molecule it produces. The frequencies of the epitopes encoded by these different polymorphisms within a single allele differ greatly. Some have a very high frequency, for example HLA-B4 and HLA-B6, and are called supratypic or ‘public’ antigens. At the other end of the scale are antigens with a very low frequency (1–2%) that are called ‘private’ antigens. Thus, epitopes with different frequencies in the population, and against which separate alloantibodies can be made, occur on a single HLA molecule and are the basis of the crossreactivity: antibodies against public antigens (also called crossreactive antigens) react with cells carrying different private antigens. Particular public antigens occur together with particular private antigens that are said to form crossreactive groups (CREGs) of (private) antigens. The higher the frequency of the public antigen in the population, the more important are antibodies against the antigen for crossreactivity. Thus, anti-HLA-B4 and -B6 are responsible for much of the crossreactivity among the HLA-B antigens.
The occurrence of crossreactive antigens is also responsible for what are called ‘splits’ of HLA antigens. Frequently, a crossreactive antigen, for example the antigen B5 in the above example, was defined before the two private antigens B51 and B52, together with which it occurs. Later, when antibodies recognizing B51 and B52 were found, the B5 antigen was ‘split’ into B51 and B52 (see Table 13.2).
Soluble HLA Class I Antigens in Plasma
Using monoclonal antibodies coated on to immunobeads and one-dimensional isoelectric focusing, followed by immunoblotting using specific class I antisera, all antigens defined to date have been detected in plasma (Doxiadis and Grosse-Wilde 1989).
Soluble HLA class I antigens (sHLA) may have important immunological effects. sHLA have been shown to inhibit alloreactive cytotoxic T cells (Zavazava et al. 1991) and to block specifically the induction of HLA alloantibody formation (Grumet et al. 1994). Allogeneic sHLA alone or complexed with antibody induces prolongation of allograft survival (Sumimoto and Kamada 1990; Wang et al. 1993a). When a graft is rejected, sHLA is shed from it and thus graft rejection episodes can be identified by serial measurements of donor-specific sHLA (Claas et al. 1993; Puppo et al. 1994).
HLA Antibodies
Mechanisms of Alloimmunization
The high density of HLA molecules on the cell surface renders allogeneic leucocytes highly immunogenic following transfusion or pregnancy. Sensitization depends upon both donor and recipient factors. Two recipient T-cell recognition mechanisms have been shown to be critical for the initiation of alloimmunity (Semple and Freedman 2002). The direct pathway occurs when recipient helper T cells interact with MHC molecules on donor APCs. The indirect pathway is more analogous to the normal immune response. Indirect recognition involves processing of allogeneic donor molecules by recipient APCs and presentation to recipient helper T cells. With indirect allorecognition, interactions between donor antigen and recipient APCs are essential for T-cell activation and subsequent antibody formation. For both pathways, the MHC molecules are expressed on the surface of the APC and are available for presentation to circulating T cells. If a T cell has a receptor with sufficient affinity for the peptide–MHC combination (first signal) and various co-stimulatory (second signal) events occur, the T cell will be activated and differentiate into an effector cell. Cytokines such as interleukin 2 (IL-2), IL-4 and alpha-interferon (IFN-α) secreted from the activated helper cells stimulate donor MHC class I-primed B cells to differentiate into the plasma cells that secrete IgG antibodies and helper T cells (Weiss and Samuelson 2003).
Development of HLA Antibodies after Transfusion
Unless measures are taken to reduce the number of transfused leucocytes (see below), a high incidence of HLA antibodies will be encountered in patients who receive multiple transfusions from different donors. However, even in subjects exposed to the blood of a single donor, the incidence of HLA antibodies is high. In a series in which patients awaiting renal grafting were given three transfusions at 2-weekly intervals from a potential donor, who in each case had a haplotype identical with one of the recipient’s haplotypes, HLA antibodies developed in some 30% of recipients (Salvatierra et al. 1980).
After the massive blood transfusion that used to be associated with open heart surgery, lymphocytotoxic antibodies and/or leucoagglutinins could be found in almost all subjects, provided that repeated tests are made, as some of the antibodies can be detected only transiently. In a series in which patients were tested at 1 week and usually also at 2, 4 and 12 weeks after open heart surgery, 52 out of 54 developed leucocyte antibodies; 12 weeks after transfusion antibodies were present in only 62.5% of the subjects (Gleichmann and Breininger 1975). The majority of HLA antibodies formed after blood transfusion are directed against class I antigens. HLA antibodies are the most important cause of antibody-mediated refractoriness to platelet transfusions (see later), of febrile transfusion reactions prior to leucoreduction and of transfusion-associated acute lung injury (see Chapter 15).
Some patients never become immunized despite repeated transfusions of blood or of platelets. Such subjects are considered to be non-responders to HLA.
Development of HLA Antibodies in Pregnancy
In primiparous women, lymphocytotoxic class I antibodies may be found as early as the twenty-fourth week of pregnancy and are present by the last trimester in 10% of women (Overweg and Engelfriet 1969). Estimates of the incidence of lymphocytotoxic antibodies after a first pregnancy vary widely: 4.3% (Ahrons 1971), 9.1% (Nymand 1974), 13% (Overweg and Engelfriet 1969) and 25% (Goodman and Masaitis 1967). The discrepancies may well be due to the varying extent of the panels of lymphocytes with which the sera were tested and the sensitivity of the techniques applied. The majority of HLA antibodies developed in pregnancy are directed against class I antigens.
Women tend to make antibodies against only certain of the HLA antigens to which they are exposed during pregnancy. In multiparous women who had had at least four pregnancies, and were therefore likely to have been exposed to antigens encoded by both of their partner’s haplotypes, the frequency of women with antibodies against only a single paternal antigen was the same as that in primiparous women (Tongio et al. 1985). Both maternal and paternal (fetal) HLA antigens play a role in the class I differential immunogenicity (Dankers et al. 2003).
Although HLA antibodies are usually IgG, they produce no obvious damage to the fetus, presumably because they are absorbed by fetal cells in the placenta.
Monoclonal HLA Antibodies
Most murine monoclonal HLA antibodies are directed against non-polymorphic determinants of the HLA molecules (Brodsky et al. 1979; Trucco et al. 1980); some antibodies detect a polymorphism that is different from those detected by alloantisera (Quaranta 1980). However, many murine monoclonals which recognize HLA antigens as defined by alloantisera have been described, particularly anti-DR and anti-DQ (Marsh and Bodmer 1989).
In addition, many human monoclonal HLA antibodies against both class I and class II antigens have now been described.
Some Features of HLA Antibodies
HLA antibodies formed after blood transfusion or pregnancy are characteristically IgG. They are complement activating and have cytotoxic properties and, like most granulocyte-reactive IgG antibodies, are leucoagglutinins (see below). HLA antibodies may be naturally occurring. Using very sensitive techniques, weak HLA antibodies, particularly anti-B8, have been demonstrated in the serum of about 1% of normal donors who had had no known pregnancies or transfusions (Tongio et al. 1985). These antibodies are usually IgM and, in the cytotoxicity test, react only with B cells, on which class I antigens are more strongly expressed than on T cells.
HLA and Haematopoietic Progenitor Cell Grafts
Graft-Versus-Host-Disease and Graft-Versus-Tumour or -Leukaemia
Haematopoietic cell (marrow, umbilical cord blood or mobilized peripheral blood) transplantation is performed to replace inadequate or defective blood cell production, for example in aplastic anaemia, sickle cell disease and thalassaemia (Walters et al. 2000; La Nasa et al. 2002; Ades et al. 2003; Atkins and Walters 2003), for adoptive immunotherapy of malignancy (Landsteiner and Levine 1929; Landsteiner 1931; Barrett 2003; Chakrabarti and Childs 2003) and for reconstitution of ‘normal’ immune function as in treatment of severe combined immunodeficiency (SCID) and Wiskott–Aldrich syndrome (Filipovich et al. 2001). The role of HLA ‘compatibility’ falls into four different areas: (1) sufficient compatibility to permit engraftment and prevent late rejection (with appropriate preparative and immunosuppressive regimens); (2) enough compatibility to minimize graft-versus-host-disease (GvHD); (3) ample immune reconstitution to permit immunosurveillance; and (4) sufficient immune potency to effect adoptive immune therapy of neoplasia. HLA identity is neither necessary nor sufficient to ensure these effects, but serological and molecular similarities are the best available surrogate assays to guide related and unrelated transplants and are a major determinant of transplant patient’s survival (Lee et al. 2007) (Figure 13.6).
Figure 13.6 Survival of patients with early, intermediate, and advanced disease depending on degree of HLA matching. (a) Early-stage disease. (b) Intermediate-stage disease. (c) Advanced-stage disease.
(Source: Lee et al. 2007. Reproduced with permission of the American Society of Hematology.)
Both GvHD and graft-versus-tumour (GvL) occur in the presence of a full HLA match, suggesting that the classical HLA molecules themselves are not targets of allosensitization, but rather present polymorphic molecules expressed by recipient cells that are recognized by the grafted immune cells.
Graft-Versus-Host Disease: the Dark Cloud of Haematopoietic Cell Grafts
Donor lymphocytes engraft, replicate and react against the normal tissues of the recipient, resulting in a syndrome known as GvHD. Myeloablative conditioning administered before transplant effectively minimizes graft rejection. The art of post-transplant immunosuppression consists of achieving a balance between graft immunocompetence and GvHD without allowing rejection of the graft. The risk of GvHD increases with genetic disparity between donor and recipient. HLA identical twins have the least chance of developing GvHD, followed by HLA identical siblings, minor degrees of mismatching among siblings, and unrelated donors of differing degrees of similarity at the MHC locus (Longster and Major 1975; Hansen et al. 1999). However, while the genetic homogeneity between donor and recipient generally decreases the risk of GvHD, it lessens the therapeutic benefit and increases the chance of tumour relapse as well (Weiden et al. 1981).
Graft-Versus-Tumor Effect: a Silver Lining of Graft-Versus-Host Disease?
One possibly beneficial effect of GvHD, or perhaps an immunological activity difficult to separate from GvHD, is rejection of recipient tumour cells by the donor immune system (graft-versus-leukaemia (GvL) effect) (Mavroudis and Barrett 1996; Mavroudis et al. 1998). The GvL effect has long been recognized to play a powerful therapeutic role in the treatment of chronic myelocytic leukaemia and more recently recognized as treatment of refractory malignant disorders including some solid tumours (graft-versus-tumour, GVT) (Childs and Barrett 2002). GVT may be the most potent form of tumour immunotherapy currently in clinical use, but its mechanism(s) of action is still poorly understood. Allogeneic T cells clearly play a fundamental role in the initiation and maintenance of the effect on neoplastic cells (Kolb et al. 1990). The risk of relapse increases markedly for patients with chronic myelogenous leukaemia who received a T-cell-depleted graft compared with a subset of patients who had received a T-cell-replete one, although the former patients avoided significant GvHD (Leak et al. 1990; Champlin et al. 2000). These results suggest that GvHD is a biological entity different from the GvL effect. In addition, upon leukaemia relapse, administration of donor lymphocyte infusion can induce clinical and molecular remission. Donor T cells may target not only tumour-specific antigens but also allelic variants of these antigens, minor histocompatibility antigens and, in the case of HLA-mismatched transplants, HLA antigens disparate from the donor but expressed by the tumour cells (Leddy and Bakemeier 1967; Lederman et al. 1983; Marijt et al. 2003). Although several theories about the mechanisms of the GvL effect have been proposed, the reasons that allogeneic T cells seem superior to native tumour immunity for some leukaemias and solid tumours remain to be clarified.
Effect of Previous Transfusion on Success of Bone Marrow Grafting
Previous transfusions, particularly from close relatives, prejudice the success of subsequent bone marrow grafting (Storb et al. 1980). The chance of rejection of the graft increases with the number of transfusions. If future recipients of a bone marrow graft need to be transfused, they should receive leucocyte-depleted blood or blood components from random donors and not from relatives. The discrepancy between the effect of transfusion of blood containing white cells on grafted bone marrow and the apparent mitigating effect on renal grafts has not been explained. The development of GvHD after the transfusion of allogeneic leucocytes is described in Chapter 15.
HLA and Organ Grafting
Renal Grafts
Significance of HLA Antibodies
When HLA alloantibodies directed against antigens expressed on the donor kidney are present in the serum of a renal graft recipient, acute or hyperacute rejection of the graft will occur. It is therefore necessary to perform a crossmatch between the patient’s serum and the B and T lymphocytes of the donor. Not all antibodies detected in the crossmatch are harmful. Cold-reacting IgM autoantibodies directed against B and T cells may be present in the serum of dialysis patients and do not appear to be harmful (Ting 1983).
Significance of Matching for HLA
The extent to which matching for HLA improves renal graft survival remains controversial. In many studies, matching has had no obvious benefit due, perhaps, to the small numbers of cases studied, interference of the many factors influencing graft survival and incomplete tissue typing. Furthermore, the survival of mismatched kidneys has improved greatly following the discovery of the beneficial effect of previous blood transfusion and of the value of ciclosporin A as an immunosuppressive drug. Nevertheless, in some large studies, a significant beneficial effect of HLA matching on long-term graft survival has been observed.
In a study in which 240 laboratories participated, the results of 30 000 first cadaver kidney transplants were analysed. Cases in which the donor and recipient were typed for all known ‘splits’ of HLA-A and -B antigens and those in which typing was restricted to the broad antigens were analysed separately. At 3 years, an 18% difference in survival rates between grafts with zero and four mismatches typed for A and B antigen splits was found, but only a 2% difference when typing was restricted to broad antigens. When A, B and DR antigens were considered together, the differences in rates of survival were 31% and 6%, respectively, in the two groups. It was concluded that typing for antigen splits is important (Opelz 1992). Molecular typing of DR alleles revealed an error rate in serological typing of about 25% (Mytelineos et al. 1990). The impact of DR matching is particularly significant if patients and donors are typed at the DNA level (Opelz et al. 1993).
In a recent study, complete matching for serologically determined HLA-A, B and DR antigens was found to have a significant and clinically important impact on short- and long-term graft survival (Opelz et al. 1999). On the other hand, partial matching provided little benefit (Held et al. 1994). The advantage of complete matching was diminished by the negative influence of longer periods of organ preservation and by the fact that in practice only 50% of perfectly matched kidneys were actually transplanted into the identified recipient. An analysis of more than 150 000 renal transplants between 1987 and 1997 in the Collaborative Transplant Study indicates that a first cadaver graft with a 6-locus mismatch has a 17% lower 10-year survival than a graft with no mismatch at these sites (Opelz et al. 1999). In the latter study, the matching effect is even more striking in patients with highly reactive preformed lymphocytotoxic antibodies. Among first cadaver transplant recipients with antibody reactivity against > 50% of the test panel, the difference in graft survival at 5 years between patients with zero or six mismatches reached 30%. Once again, correction of serological HLA typing errors by more accurate DNA typing resulted in a significantly improved HLA matching effect, and matching for the class II locus HLA-DP, a locus that can be typed reliably only by DNA methods, showed a significant effect for cadaver kidney re-transplants.
Non-HLA transplant immunity may be more important for long-term graft survival (Opelz et al. 2005).
For brief discussions of the importance of ABO as a major histocompatibility system and of the possible effects of Lewis groups on renal transplantation, see Chapter 4.
Liver Grafts and Heart–Lung Grafts
The survival of liver grafts is reportedly improved with HLA matching and is worse when the T-cell crossmatch is positive (Nikaein et al. 1994). However, this finding could not be confirmed in the Collaborative Transplant analysis (Opelz et al. 1999). Matching for HLA-DR diminished the frequency of rejection episodes after heart transplantation from 34% to 16%, and at 3 months there was an additional beneficial effect of HLA-B matching (Sheldon et al. 1994). An independent study of heart transplants showed a highly significant impact of HLA compatibility on graft outcome (P < 0.0001) (Opelz et al. 1999). In practice, matching for HLA is much more difficult in the transplantation of liver and heart than of kidney, mainly because there is no large pool of HLA-typed recipients to choose from.
HLA-Antibody-Mediated Rejection of Solid Organs
Antibody-mediated hyperacute rejection was the first rejection phenotype observed in human solid organ transplants. Hyperacute rejection was all but eliminated by using reliable antibody crossmatch technologies. Since then, the focus was on T-cell mediated rejection and de novo donor-specific antibodies were considered an epiphenomenon of cognate T-cell activation. The immune theory was that controlling the T-cell response would entail elimination of antibody-mediated rejection (ABMR). With modern immunosuppressive drugs, T-cell-mediated rejection is essentially treatable. However, this did not prevent ABMR from emerging as a significant phenotype in all types of organ transplants. It became obvious that both rejection types require distinct treatment and thus reliable diagnosis. This is the current challenge. ABMR, depending on stage, grade, time course, organ type or prior treatment, can present with a wide spectrum of phenotypes. This review summarizes the current diagnostic consensus for ABMR, describes unmet needs and challenges in diagnostics, and proposes new approaches for consideration. Antibody-mediated kidney allograft rejection (AMR) has come into the focus of transplant immunologists. Intravenous immunoglobulin, rituximab, bortezomib, and eculizumab have been used to treat patients with acute AMR, apart from the standard treatment of antibody removal with plasma exchange or immunoadsorption and steroid pulses.
Immunomodulatory Effect(S) of Transfusion
As knowledge about the mechanisms of immune responsiveness and tolerance evolves, and as tools to measure alterations in immunity become available, additional immunological consequences of blood transfusion are being detected. Numerous variations in circulating blood cells have been reported in patients transfused with allogeneic blood (see below). Some of these changes persist for months or even longer after transfusion. The lingering question has been whether these observations represent no more than laboratory curiosities, or whether they reflect some clinically relevant alteration in the recipient’s immune status, the so-called ‘immunomodulatory effects’ of blood transfusion. Based on the sum of clinical evidence (see below) immunomodulation seems likely to be added to the list of unintended consequences of allogeneic blood transfusion. The magnitude and importance of these effects, the causative agents, the biological mechanisms and the patients or patient groups that are at particular risk have yet to be defined (Klein 1999).
Dzik (2003) has suggested that there may be two categories of immunosuppressive transfusion effect: one that is HLA dependent and directed against adaptive immunity and a second that is mild, non-specific and directed against innate immunity. The non-specific effect might result from the infusion of blood cells that undergo apoptosis during refrigerated storage. The infusion of apoptotic cells has been shown to be immunosuppressive in animal models. Immunosuppression resulting from the infusion of apoptotic cells may be linked to transforming growth factor beta (TGF-β) (Dzik 2003).
Changes in Recipient’s Lymphocytes after Blood Transfusion
Following the transfusion of large amounts of fresh or stored blood, changes develop in the recipient’s lymphocytes after an interval of about 1 week. Atypical lymphocytes increase by a factor of five or more and lymphocytes may incorporate 3H-thymidine in vitro at an increased rate. Values return to the pretransfusion level by about 3 weeks. Changes are not seen after transfusion of frozen and washed (leucocyte-depleted) red cells (Schechter et al. 1972). Confirmatory observations were published by Hutchinson and co-workers (1976). The changes are interpreted as a response to donor HLA antigens (presumably of the Dw series) and may be regarded as those of an MLC in vivo. A number of other alterations of immune cells including a decrease in NK function and delayed hypersensitivity have been published (Tartter et al. 1986, 1989; Jensen et al. 1992). Blood transfusion alters immune cell antigen expression in premature neonates and may initially be immunostimulatory and later immunosuppressive (Wang-Rodriguez et al. 2000). Donor lymphocytes may circulate for prolonged periods in some patient groups, such as trauma victims, whereas in others such as patients infected with HIV, microchimerism appears to be transient (Kruskall et al. 2001; Lee et al. 1995, 1999, 2001). The immunomodulatory role of persistent microchimerism post transfusion and its relationship to the HLA system are areas of active investigation. For the relationship of microchimerism and transfusion-associated GVHD, see Chapter 15.
Effect of Previous Transfusion on Success of Renal Graft
Patients who have antibodies against HLA antigens of the donor undergo acute rejection of renal allografts. On the other hand, blood transfusion has been shown to have a striking effect in improving the survival of subsequent renal grafts in subjects who have not developed cytotoxic antibodies, or who have done so and have received renal grafts from HLA-compatible donors (Opelz et al. 1973; van Hooff et al. 1976). Leucocytes in the donor blood have been found to be essential for the beneficial effect (Persijn 1984).
After the introduction of more potent immunosuppression with ciclosporin, transfusions before cadaver grafting were found to confer little additional benefit (Kaban et al. 1983; Lundgren et al. 1986; Opelz 1987), although a single-centre randomized study attributed pretransplant transfusion benefit to reduced mortality related to immunosuppression (Vanrenterghem et al. 1994). However, transfusions improved the 1-year graft survival rate by 8% (P < 0.01) in recipients of a one-DR mismatched graft and by 10% (P < 0.01) in recipients of a two-DR mismatched graft (Iwaki et al. 1990). Two to four transfusions from random donors were sufficient to obtain this effect. This study concluded that despite the use of ciclosporin, the practice of giving deliberate transfusions before grafting should not be abandoned. In a randomized, controlled multicentre trial, cadaver graft survival rate was significantly higher in the 205 recipients who underwent three pretransplant transfusions than in the 218 patients who did not receive transfusions (Opelz et al. 1997).
When a kidney from a live donor is used, it is possible to give both transfusion and graft from the same donor. Donor-specific blood transfusions (DSTs) lead to increased graft survival rates (Salvatierra et al. 1981, 1986; Kaplan 1984). A disadvantage of DST is that the patient may develop lymphocytotoxic antibodies against donor HLA antigens. In animal models it was found that heat treatment of the donor blood (Martinelli et al. 1987), or pre-treatment of the recipient with donor leucocytes coated with antilymphocyte antibody, diminished the chance of such immunization (Susal et al. 1990). Treatment of the patient with azathioprine also had this effect (Anderson et al. 1982).
Several mechanisms have been suggested to explain the beneficial effect of previous blood transfusions on renal graft survival: (1) the induction of increased suppressor cell activity (Marquet and Heystek 1981; Quigley et al. 1989); (2) decreased natural killer cell activity (Gascon et al. 1984); (3) specific unresponsiveness due to idiotype antibodies, which inactivate T-cell clones (Woodruff and van Rood 1983; Kawamura et al. 1989); (4) impairment of the function of the mononuclear phagocyte system (MPS) by iron loading (Akbar et al. 1986; de Sousa 1989); (5) deletion of clones of cells, which are first activated by blood transfusion and then killed or inactivated by high dose immunosuppressive therapy during the anamnestic response after transplantation (Terasaki 1984); and (6) the production of non-cytotoxic, Fc receptor-blocking antibodies (MacLeod et al. 1985; Petranyi et al. 1988).
Sharing of MHC antigens between donor and recipient has been found to determine the extent of the blood transfusion effect. The survival of kidney grafts in recipients who were given transfusions, and who shared one HLA-DR antigen with the donors, was significantly better (81% at 5 years) than in recipients who were given transfusions from donors mismatched for both DR antigens (57% at 5 years), or in recipients who were not transfused (45%). Immunization occurred less frequently in the recipients who shared one DR antigen with the donor (Lagaaij et al. 1989). In another study, sharing of one HLA haplotype (or at least one HLA B and DR antigens) between donor and recipients had a mitigating effect because it led to a specific suppression of the formation of cytotoxic T lymphocytes (CTLs), i.e. to CTL non-responsiveness; recipients of blood from fully identical donors remained CTL responders (van Twuyver et al. 1991). Furthermore, transfusion of blood from HLA-identical donors induces the generation of suppressor cell independent, high-affinity CTL against donor antigens (van Twuyver et al. 1994). These mitigating and immunizing effects are donor specific, but the beneficial effect of blood transfusion on kidney survival is also due to a non-specific effect. Transfusion of blood from donors who share one HLA haplotype induces a general decrease in the usage of T-cell Vβ families (Munson et al. 1995). These effects are probably due to the survival of donor lymphocytes in the recipient. Studies in mice have shown that sharing of H2 antigens between donor and recipient of a blood transfusion facilitates the persistence of donor lymphoid cells in the recipient, which is associated with tolerance for donor alloantigens. Donor lymphocytes can be detected 10–20 years after transplantation in patients in whom the graft survives for long periods of time. Such chimerism may be important in modulating the immune response (Starzl et al. 1992).
Effect of Transfusion on Tumour Growth and Recurrence of Cancer
A retrospective analysis of the recurrence rate of carcinoma of the colon after surgical resection first suggested that the 5-year disease-free survival rate was reduced by blood transfusion given at the time of surgery (Burrows and Tartter 1982). However, despite numerous subsequent reports, including more than 100 observational studies and three controlled trials, and several meta-analyses, the relationship between blood transfusion, cancer growth and cancer-free survival remains murky and contradictory (Klein 2001). The numerous variables including different tumours, locations, extent of disease, histological grade and modes of treatment make this a particularly difficult area to evaluate.
Prospective randomized clinical studies have been conducted with patients undergoing surgery for colorectal carcinoma. In one large multicentre randomized trial, patients who were operated on because of colorectal cancer and who needed blood transfusion were randomized to an autologous or allogeneic transfusion regimen. At study conclusion, patients received allogeneic blood only (133), autologous and allogeneic blood (66), autologous blood only (112) or no blood at all (164). There were no significant differences between the groups receiving allogeneic blood or autologous blood only: at 4 years, survival rates were 67% and 62%, respectively, and in survivors, no recurrence of cancer in 63% and 66% respectively. On the other hand, many patients did not receive the ‘treatment’ specified by their prospective treatment assignment, and cancer recurred significantly more frequently in the transfused than in the non-transfused patients. This difference may have been associated with the circumstances that necessitated transfusion (Klein 1999). The red cells transfused in the allogeneic arm were buffy coat depleted as was the routine in the Netherlands at the time. In a second controlled study of colorectal cancer in the Netherlands, leucoreduced red cells were compared with buffy coat-reduced red cells. No significant differences were found between the two trial transfusions in survival, disease-free survival or cancer recurrence rate after an average follow-up of 36 months. Patients who had a curative resection and who received blood of any sort had a lower 3-year survival than non-transfused patients (69% vs. 81%, P = 0.001). These observations confirm an association between blood transfusion and poor patient survival, but suggest that the relation is not due to promotion of cancer (Houbiers et al. 1994). The third prospective study of colorectal cancer from a single centre in Germany found that blood transfusion was an independent factor associated with tumour recurrence, and that survival of transfused patients tended to be shorter although the difference was not statistically significant (Heiss et al. 1994). There may well be some subset of patients, perhaps defined by immune status or tumour subtype, that is particularly susceptible to the effects of allogeneic transfusion. Demonstration of such a difference will probably require a large, carefully controlled, prospective study.
Effect of Transfusion on Postoperative Infections
As is the case with cancer, a large number of observational studies find an association between allogeneic blood transfusion and postoperative bacterial infection, while a few do not (Vamvakas and Blachman 2001). For example, Carson and co-workers (1999) conducted a retrospective cohort study of 9598 consecutive patients with hip fracture who underwent surgical repair between 1983 and 1993 at 20 hospitals across the USA. Bacterial infection, defined as bacteraemia, pneumonia, deep wound infection or septic arthritis/osteomyelitis was the primary endpoint and numerous variables were included in the statistical model; a highly significant association was found between serious postoperative infection and transfusion. Chang and co-workers (2000) analysed a database of 1349 patients undergoing elective colorectal surgery for any disease of the colon or rectum at 11 university hospitals across Canada. To adjust for confounding effects associated with remote infections such as pneumonia and urinary tract infections, the study limited the analysis to postoperative wound infection. Allogeneic blood transfusion was a highly significant independent predictor of postoperative wound infection. Vamvakas and Carven (1998) reported a retrospective cohort study of 416 coronary artery bypass graft patients admitted to one hospital. The endpoints were limited to postoperative wound infection or pneumonia, and adjustment was made for the effects of chronic systemic illness and specific risk factors for wound infection or pneumonia. The risk of postoperative wound infection or pneumonia increased by 6% per unit of allogeneic red blood cells (RBCs) and/or platelets transfused, or by 43% for a patient receiving the mean transfusion dose of 7.2 units of either component. Nevertheless, these analyses are inevitably flawed, despite meticulous multivariate testing, by the numerous variables that predispose to postoperative infection (comorbidities, catheters, respirator time, impaired consciousness, etc.), not to mention factors related to the blood components such as storage time and method of preparation (Vamvakas and Carven 1998).
Seven randomized controlled trials compare the incidence of postoperative infection between recipients of buffy coat-reduced (Heiss et al. 1993; Busch et al. 1994; Houbiers et al. 1994; Jensen et al. 1996; van de Watering et al. 1998) or standard allogeneic red cells (Tartter et al. 1998) or whole blood (Jensen et al. 1992) and recipients of autologous or WBC-reduced, buffy coat-reduced allogeneic red cells or whole blood. Two studies (Jensen et al. 1992, 1996) reported a significant effect, two studies (Heiss et al. 1993; van de Watering et al. 1998) reported a marginal effect, and three studies (Busch et al. 1994; Houbiers et al. 1994; Tartter et al. 1998) did not detect an effect. The strengths and weakness of these studies have been analysed exhaustively (Vamvakas and Blachman 2001). However, insufficient data are available to perform the kind of meta-analysis that might help draw conclusions from these studies.
Postoperative Mortality
In addition to the possible association between allogeneic transfusion and postoperative infection, van de Watering and co-workers (1998) detected an unexpected association between WBC-containing allogeneic blood transfusion and postoperative mortality from causes other than postoperative infection. In total, 24 out of 306 patients (7.8%) transfused with buffy coat-reduced red cells died, compared with 11 out of 305 patients (3.6%) receiving buffy coat-reduced red cells that were leucoreduced before storage, and 10 out of 303 patients (3.3%) receiving buffy coat-reduced red cells that were leucoreduced after storage (P = 0.015). The overall difference in 60-day mortality was due to a highly significant difference among the three randomization arms. The number of RBC units transfused was the most significant predictor of postoperative mortality. The association between leucocyte-containing allogeneic blood and increased mortality may be limited to cardiac surgery and should not be extended to other clinical settings. At the very least, the finding requires confirmation by a study designed with mortality as the primary endpoint.
Possible Role of HLA in Habitual Abortion
Parental sharing of HLA antigens has been thought to be a cause of habitual abortion. In such cases, non-cytotoxic antibodies that are normally produced in the mother and that protect the fetus are absent (Adinolfi 1986; Scott et al. 1987).
Immunization of women with leucocytes has been employed with the object of correcting the immunological unresponsiveness (Taylor and Falk 1981; Beer et al. 1985). One problem in assessing the benefit of such immunization is that the definition of habitual abortion varies. Furthermore the chance of a successful pregnancy after three abortions is about 60% (Regan 1991). One prospective randomized trial (Mowbray et al. 1987) has shown an apparent benefit; in another trial, no clear advantage of leucocyte injection was observed, and the authors expressed their concern about severe growth retardation seen in some fetuses (Beer et al. 1985). The Recurrent Miscarriage Study enrolled women who had had three or more spontaneous abortions of unknown cause in a double-blind, multicentre, randomized clinical trial (Ober et al. 1999). In total, 91 women were assigned to immunization with paternal mononuclear cells (treatment) and 92 to immunization with sterile saline (control). The primary endpoints were the inability to achieve pregnancy within 12 months of randomization, or a pregnancy that terminated before 28 weeks of gestation (failure); and pregnancy of 28 or more weeks of gestation (success). Immunization with paternal mononuclear cells did not improve pregnancy outcome in women with unexplained recurrent miscarriage. However, it is possible that a subset of responders might be identified by using some as yet unrecognized laboratory determination or susceptibility factor. Until this is possible, immune therapy in women with habitual abortion should be restricted to clinical trials (Moloney et al. 1989).
Tests for HLA Alleles, Antigens and Antibodies
HLA Alleles
HLA alleles can be determined directly at the DNA level. The resolution of DNA-based typing is limited only by the available allele-specific probes. The relevant techniques are based on several different principles (see Chapter 3). While the heterogeneity of the MHC has made high-resolution typing problematic for matching donor and recipient for transplantation (Petersdorf et al. 2001), the stringency of the HLA role in antigen presentation has made high resolution increasingly desirable for immunotherapy trials.
Anthony Nolan HLA informatics group publishes up-to-date online HLA Class I and II Sequence Alignments (www.anthonynolan.com/HIG/data.html).
Sequence-Specific Oligonucleotides
DNA is amplified in the polymerase chain reaction (PCR) and a set of sequence-specific oligonucleotides (SSOs) is used in a dot-blot or reverse dot-blot hybridization technique to detect allelic sequences (Saiki et al. 1986, 1989; Ng et al. 1993). Several modifications of this technique have been described (Bidwell 1994). In one, a strand of heat-denatured, amplified DNA is ligated with SSOs by an added ligase. Ligation only occurs when the sequences of the DNA and SSO are identical. The ligated product is detected by enzyme-linked immunosorbent assay (ELISA) (Fischer et al. 1995). The techniques permit the identification of alleles, even of those differing from each other by a single nucleotide.
Sequence-Specific Primers
PCR is performed with a set of sequence-specific primers (SSPs) that will only amplify DNA with sequences complementary to the primers (Olerup et al. 1993; Olerup 1994; Bunce et al. 1995). A simple and quick SSP test in microplates has been described (Chia et al. 1994).
The limitations of both SSO and SSP are requirements for a large number of PCRs to include the known alleles, and the inability to identify polymorphisms unless the variation happens to lie within the region spanned by the assay. These limitations are addressed by nucleotide sequencing of PCR-amplified DNA, the method of choice for ‘high-resolution’ typing that is required in the selection of an unrelated stem cell donor (Spurkland et al. 1993). High-throughput robotic sequence-based typing allows daily sequencing of hundreds of genomic fragments, and high-density array technology promises to permit extensive typing of polymorphisms, both known and unknown, on microchips (Adams et al. 2001; Wang et al. 2003).
HLA Antigens and Antibodies
Class I antigens are determined using the lymphocytotoxicity test. Crossreactivity and the lack of specific antisera led to difficulties in HLA typing; several antisera must be used in typing for a particular antigen. In serological typing for DR and DQ antigens, the two-colour fluorescence test or the lymphocytotoxicity test on B cells is applied (see below). These same techniques are used for the detection of class I and class II antibodies respectively. Lymphocytotoxicity is declining in interest in the USA as most laboratories switch to easier, higher resolution molecular methods. However, immunological methods remain valuable to characterize functional aspects of HLA as molecular methods cannot define whether an HLA allele is expressed or how a sequence correlates with empirically determined antigen importance.
Lymphocytotoxicity Test
Complement-dependent cytotoxicity remains the standard test for determining HLA class I antigens. Lymphocytes are incubated with antibody and rabbit complement, and a dye (Trypan blue or eosin) is then added. If the lymphocytes carry an antigen corresponding to the antibody, complement is fixed, the cell membrane is damaged and dye enters and stains the cell (blue or red). The percentage of stained cells is counted. Live cells are unstained, smaller and refractile. It is essential to use a pure lymphocyte suspension, as platelets carry A, B and C antigens and granulocytes are always killed in the cytotoxic assay and stain non-specifically. Details of the NIH-recommended lymphocytotoxicity test, using microdroplets, were published by Terasaki and co-workers (1973).
For the determination of DR and DQ antigens by lymphocytotoxicity, B lymphocytes can be isolated: (1) by removing the T lymphocytes from a lymphocyte suspension by rosetting with Z-aminoethylisothiouronium bromide-treated sheep red cells and centrifugation on Ficoll-hypaque (density 1.077) (Pellegrino et al. 1976); (2) by the use of nylon fibre columns (Wernet et al. 1977); or (3) by the use of magnetic beads coated with monoclonal antibodies specific for class II epitopes (Vartdal et al. 1986).
In the two-colour fluorescence tests the IgG on the B cells is capped with FITC-labelled anti-IgG followed by a cytot*oxicity test. The B cells can be distinguished from the T cells by the green IgG cap on their surface. For a description of the technique, see van Rood and van Leeuwen (1976).
The Mixed Lymphocyte Culture
This test was described by Bain and co-workers (1964). The principle is to irradiate or add a substance such as mitomycin C to one sample, usually the donor’s, and to mix these lymphocytes with those from another subject such as a potential recipient. Irradiation, or treatment with mitomycin C, prevents lymphocytes from transforming to blast cells but does not destroy their ability to stimulate other lymphocytes. Blast transformation of the untreated lymphocytes indicates that they have recognized a foreign antigen on the treated lymphocytes (Bach and Voynow 1966), and this transformation can be assessed by measuring the incorporation of tritiated thymidine (one-way MLC).
In the MLC, the cells that stimulate are B cells and monocytes carrying Dw determinants and class II antigens. Those that respond are T cells (Potter and Moore 1977).
If irradiated or mitomycin C-treated stimulator cells, homozygous for a Dw determinant (homozygous typing cells, or HT), are used they can only stimulate untreated lymphocytes that do not carry the Dw determinant for which they are homozygous. Thus, panels of HTC are used to identify Dw determinants (Bradley et al. 1972).
The two-way MLC, in which the lymphocytes in both samples are able to respond by blast formation, has been used as a final test for HLA identity of donors and recipients of bone marrow who are serologically identical.
HLA Antibody Detection
Typed repository cell lines are used in a complement-dependent cytotoxicity assay to identify alloantibodies in sera of sensitized subjects. The percentage of cell lines killed by the sera is used as a rough measure of the degree of sensitization or ‘panel reactive antibody’ (PRA) reactivity. Some antibodies activate complement, yet kill cells inefficiently, a phenomenon known as ‘cytotoxicity negative absorption positive’ (CYNAP) (Lublin and Grumet 1982). The CYNAP phenomenon may result in underestimation of sensitization. However, augmentation of the assay to increase sensitivity may implicate innocuous antibodies and thus over-estimate clinically relevant sensitization. Another method of identifying alloantibodies uses (Mollicone et al. 1988) flow cytometry of a variety of microbeads loaded with known HLA alleles (Guertler et al. 1984; Moses et al. 2000). An interlaboratory comparison of techniques suggests that considerable inconsistencies in serum screening and crossmatching exist among laboratories participating in the American Society for Histocompatibility and Immunogenetics/College of American Pathologists surveys (Duquesnoy and Marrari 2003). The lack of uniformity in test results may limit the usefulness of these methods in a clinical setting.
Other Antigens Found on Leucocytes
Some red cell antigens are also found on leucocytes; see Chapters 4 and 6.
Antigens Located on Granulocytes (Human Neutrophil Antigens)
Nomenclature: Confusing, Controversial and Evolving
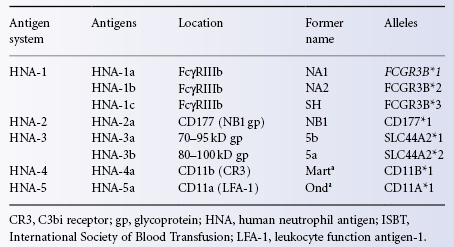
Neutrophil antigens were first characterized using sera collected from neutropenic patients who formed clinically important allo- and autoantibodies. Although the presence of the first granulocyte-specific antigen, NA1, was inferred from the presence of an antibody in a case of neonatal neutropenia in 1960, the antigen was not identified until 1966 (Lalezari et al. 1960; Lalezari and Bernard 1966a). As new antigens were discovered, nomenclature threatened to assume some of the quirky randomness that characterized red cell blood group antigens. A new nomenclature was proposed in 1998 to permit separate notations for the phenotype associated with the glycoprotein location and for the alleles, according to the guidelines for human gene nomenclature (Bux 1999). The proposed nomenclature has been criticized for including antigens found on cells other than granulocytes, however it is now generally accepted. The antigen systems are referred to as human neutrophil antigens (HNA). The antigen systems, the polymorphic forms of the immunogenic proteins, are indicated by integers and specific antigens within each system are designated alphabetically by date of publication. Alleles of the coding genes are named according to the Guidelines for Human Gene Nomenclature. Neutrophil antigens NA1 and NA2 became HNA-1a and HNA-1b in the new nomenclature and the third antigen reported, NB1 became HNA-2a (Table 13.1).
Table 13.1 ISBT Human Neutrophil Antigen (HNA) nomenclature.
Source: Adapted from Wang et al. 2009. Reproduced with permission of Elsevier.
The HNA-1 System
HNA-1a, -1b and -1c Polymorphisms
The HNA-1 antigen system consists of the three alleles HNA-1a, -1b and -1c (Bux et al. 1997). The antigens, also known as NA1, NA2 and SH, are expressed only on neutrophils and expression varies depending upon the maturity of the cell (Table 13.1). The gene frequencies of the three alleles vary widely among different racial groups (Hessner et al. 1999; Matsuo et al. 2001). Among white subjects, the frequency of the gene encoding HBA-1a, FCGR3B*1, is between 0.30 and 0.37, and the frequency of the gene encoding HNA-1b, FCGR3B*2, is from 0.63 to 0.70. In Japanese and Chinese populations, the FCGR3B*1 gene frequency is from 0.60 to 0.66, and the FCGR3B*2 gene frequency is from 0.30 to 0.33. The frequency of the gene encoding HNA-1c, FCGR3B*3, also varies among racial groups. FCGR3B*3 is expressed by neutrophils from 4% to 5% of white subjects and 25 to 38% of African Americans (Kissel et al. 2000).
The FCGR3B*1 gene differs from the FCGR3B*2 gene by only five nucleotides in the coding region at positions 141, 147, 227, 277 and 349 (Ory et al. 1989; Ravetch and Perussia 1989; Huizinga et al. 1990; Trounstine et al. 1990). Four of the nucleotide changes result in changes in amino acid sequence between the HNA-1a and the HNA-1b forms of the glycoprotein. The fifth polymorphism at site 147 is silent. The glycosylation pattern differs between the two antigens because of the two nucleotide changes at bases 227 and 277. The HNA-1a form of FcγRIIIb has six N-linked glycosylation sites and the HNA-1a form has four glycosylation sites.
The gene encoding the HNA-1c form of FγcRIIIb, FCGR3B*3, is identical to FCGR3B*2 except for a C-to-A substitution at amino acid 78 of FcRIIIb (Bux et al. 1997). In many cases, FCGR3B*3 exists on the same chromosome with a second or duplicate FCGR3B gene (Koene et al. 1998).
Several other sequence variations in FCGR3B have been described. These chimeric alleles have single-base substitutions involving one of the five SNPs that distinguish FCGRB3B*1 and FCGR3B*2. FCGR3B alleles that more closely resembled FCGR3B*2 were found more often in African Americans than in white people or Japanese people (Matsuo et al. 2001).
Function of HNA-1 Antigens
The low-affinity FcγRIIIb receptors link humoral and cellular immune function. The FcγRIIIb on effector cells bind cytotoxic IgG molecules and immune complexes containing IgG. Polymorphisms in FcγRIIIb affect neutrophil function. Neutrophils that are homozygous for HNA-1a have a greater affinity for IgG3 than do those that are homozygous for HNA-1b (Nagarajan et al. 1995). Neutrophils from subjects homozygous for HNA-1b phagocytize erythrocytes sensitized with IgG1 and IgG3 anti-Rh monoclonal antibodies and bacteria opsonized with IgG1 less efficiently than do granulocytes homozygous for HNA-1a (Bredius et al. 1994).
FcγRIIIb Deficiency
Blood cells from patients with paroxysmal nocturnal haemoglobinuria (PNH) lack the GPI-linked glycoproteins and their granulocytes express reduced amounts of FcγRIIIb and the HNA-1 antigens (Huizinga et al. 1990). Genetic deficiency of granulocyte FcγRIIIb and the HNA-1 antigens has been reported. With inherited deficiency of FcγRIIIb, the FCGR3B gene is deleted along with an adjacent gene, FCGR2C (De Haas et al. 1995). Despite the primary role of FcγRIIIb in neutrophil function, deletion of the entire FcγRIIIB gene results in no obvious clinical abnormality. Although most subjects who lack FcγRIIIb appear healthy, too few have been studied to ensure that no subtle alteration in immune function is present. In a study of 21 FcγRIIIb subjects with FcγRIIIb deficiency, two were found to have autoimmune thyroiditis and four had sustained multiple episodes of bacterial infections (De Haas et al. 1995).
FCGR3B Polymorphisms and Disease Associations
Several studies suggest that FCGR3B polymorphisms are associated with autoimmune and inflammatory disorders. Children with chronic immune thrombocytopenic purpura were more likely to be FCGR3B*1 homozygous than were control subjects (Foster et al. 2001), whereas Spanish patients with systemic lupus erythematosus were more likely to be FCGR3B*2 homozygous (Gonzalez-Escribano et al. 2002). Myasthenia gravis is reportedly more severe in FCGR3B*1 homozygous patients (Raknes et al. 1998), but multiple sclerosis may take a more benign course (Myhr et al. 1999). Patients with chronic granulomatous disease who are FCGR3B*1 homozygous are less likely to develop major gastrointestinal or urinary tract infectious complications compared with those who are heterozygous or FCGR3B*2 homozygous (Foster et al. 1998). As FCGR3B is clustered with FCGR3BA and FCGR2B on chromosome 1q22, some of these findings may reflect in part linkage disequilibrium among Fc receptors.
The HNA-2 System
The HNA-2 system has a single allele, HNA-2a (NB1) expressed only on neutrophils, metamyelocytes and myelocytes (Stroncek et al. 1998a). The 58- to 64-kDa glycoprotein that carries HNA-2a (NB1 gp) is located on neutrophil plasma membranes and in secondary granules, and is linked to the plasma membrane by a glycosylphosphatidylinositol (GPI) anchor. HNA-2a is expressed on 45–65% of circulating neutrophils; expression is greater on neutrophils from women (Stroncek et al. 1996; Matsuo et al. 2001). Pregnant women express HNA-2a more strongly than do healthy female blood donors (Caruccio et al. 2003). Expression of HNA-2a decreases with age in women, but remains constant in men. Administration of G-CSF to healthy subjects can increase the proportion of neutrophils expressing HNA-2a to near 90% (Stroncek et al. 1998b). Monoclonal antibodies specific to HNA-2a have been clustered as CD177. The role of CD177 in neutrophil function is unknown. Women who lack NB1 gp are healthy. Although the expression of HNA-2a is reduced on neutrophils from patients with PNH and chronic myelocytic leukaemia (CML), no clinical significance has been attributed to this observation.
HNA-2 Polymorphisms
HNA-2a is expressed on neutrophils by approximately 97% of whites, 95% of African Americans and 89–99% of Japanese (Matsuo et al. 2000; Taniguchi et al. 2002). HNA-2a has been reported to have an allele, NB2, but the product of this gene cannot be detected consistently with alloantisera, and no monoclonal antibody has been raised for NB2 (Stroncek et al. 1993a). The HNS-2a-negative neutrophil phenotype is due to a CD177 transcription defect. HNA-2a genes from two women with HNA-2a-negative neutrophils, who produced HNA-2a-specific alloantibodies have been studied and CD177 cDNA sequences were present in both women. Both women were clinically unaffected.
The HNA-3 System
The HNA-3a and -3b antigens derive from nucleotide polymorphisms in the choline transporter-like protein-2 gene (SLC44A2) on chromosome 4 that leads to a variations (461 G>A; Arg154Gln) at amino acid position 154 (Curtis et al. 2010; Greinacher et al. 2010). The resulting protein, reacts with HNA-3a-specific antibodies and is recognized as the HNA-3a antigen. The HNA-3a antigen was previously known as 5b.Potent anti-HNA-3a agglutinins in transfused plasma can cause transfusion-related acute lung injury (TRALI) (see Chapter 15) .Molecular identification of this antigen has led to development of assays to screen blood donors and help diagnose TRALI. HNA-3a is expressed by a variety of cells including neutrophils, lymphocytes, platelets, endothelial cells, kidney, spleen and placental cells (van Rood and Ernisse 1968) and weakly expressed on red cells (Rosenfield et al. 1967).
HNA-4 and HNA-5 Systems
The HNA-4 and HNA-5 antigens are located on the β2 integrins. Each system contains only a single antigen, HNA-4a and HNA-5a. The HNA-4a antigen, previously known as Marta, has a phenotype frequency of 99.1% in whites (Kline et al. 1982). HNA-4a is expressed on granulocytes, monocytes and lymphocytes, but not on platelets or red blood cells. HNA-4a has been located on the αM chain (CD11b) of the receptor CR3. Neonatal alloimmune neutropenia has been caused by antibody to HNA-4a, but this is the exception rather than the rule (Fung et al. 2003). A second polymorphism of the β2 integrins, HNA-5a, previously Onda with a frequency of 95%, is located on the α-chain of the leucocyte function-associated antigen (LFA-1, CD11a) molecule (Simsek et al. 1996).
Other Granulocyte-Specific Antigens
Other granulocyte-specific antigens are ND1, NE1 and LAN (Lalezari and Radel 1974; Verheugt et al. 1978; Claas et al. 1979; Rodwell et al. 1991). Like the NA antigens, LAN is located on the FcγRIIIb (Metcalfe and Waters 1992). Another high-frequency antigen, also located on the FcγRIIIb, was described by Bux and colleagues (1994). The antigen NC1 (Lalezari and Radel 1974) is identical with HNA-1b (Bux et al. 1995a). Most granulocyte-specific antigens have been defined by alloantibodies, but ND1 and NE1 were defined by autoantibodies. These granulocyte-specific antigens appear to be true differentiation antigens, as they appear at the myelocyte or metamyelocyte stage or even later (Lalezari 1977; Evans and Mage 1978).
Antigens on Granulocytes and Monocytes
The following antigens have been shown to be present on granulocytes and monocytes: HGA-1 (Thompson et al. 1980) and the HMA-1 and HMA-2 antigens, products of a biallelic gene (Jager et al. 1986). The AYD antigen is shared by granulocytes, monocytes and endothelial cells (Thompson and Severson 1980). The 9a antigen, which was first thought to be granulocyte specific, is also expressed on monocytes (Jager et al. 1986).
Antibodies to Neutrophil Antigens
Antibodies to neutrophil antigens may be responsible for five different clinical syndromes: (1) neonatal allo- and isoimmune neutropenia; (2) autoimmune neutropenia; (3) transfusion-related alloimmune neutropenia; (4) pulmonary infiltrates following transfusion (TRALI); and (5) febrile reactions following transfusion. The last two conditions are discussed in detail in Chapter 15. Transfusion-related alloimmune neutropenia is probably a variant of TRALI and appears to be rare (Wallis et al. 2002).
Neonatal Alloimmune Neutropenia (NAIN)
This syndrome, analogous to haemolytic disease of the newborn (HDN), is usually recognized when infection in a newborn infant is found to be accompanied by severe neutropenia. Neutrophil antigens in the fetus that are inherited from the father but foreign to the mother provoke formation of maternal IgG antibodies that cross the placenta and react with the neonate’s neutrophils (Lalezari and Bernard 1966b). Absolute neutrophil counts typically range from 0.100 to 0.200 × 109/l. Bone marrow examination reveals myeloid hyperplasia. The syndrome is self-limited, but may persist from days to weeks as passive antibody is cleared (Bux et al. 1992). NAIN is likely under diagnosed. Symptoms include delayed separation of the cord and mild skin or respiratory infections and are frequently mild and non-specific. Severe and overwhelming infection can occur and the mortality rate for NAIN is about 5%. Treatment with IVIG or recombinant cytokines such as granulocyte colony-stimulating factor (G-CSF) has met with variable success (Maheshwari et al. 2002).
In reviewing the syndrome, Lalezari and Radel (1974) described results in 19 infants from 10 families. The specificity of the antibody in three families was anti-HNA-1a; in two, anti-HNA-1b; in four, anti-HNA-2a; and in one, not determined. In four of the families the first-born infant was affected. Antibodies of other specificities have been implicated, but much less frequently (Stroncek 2002).
A prospective survey of some 200 pregnant women, either primiparae at term or multiparae, in which the woman’s serum was tested against her partner’s granulocytes and lymphocytes, indicated that the incidence of neutrophil antibodies was about 3% (Verheugt et al. 1979). A prospective study of unselected pregnancies found a NAIN incidence of 0.81% based on screening 247 ‘full term’ cord blood samples for neutropenia and the presence of granulocyte antibodies (Williams and Fung 2006).
Neonatal Isoimmune Neutropenia (NIN)
Subjects who lack a membrane glycoprotein due to deletion of the gene encoding the glycoprotein may form strong antibodies (named isoantibodies) against non-polymorphic determinants on the glycoprotein. Severe neonatal neutropenia caused by maternal isoantibodies may occur in infants from mothers with a deletion of the FcγRIIIb gene (Huizinga et al. 1990; Stroncek et al. 1991; Cartron et al. 1992; Fromont et al. 1992). NIN is usually self-limited.
Autoantibodies to Granulocytes
The first convincing case that implicated autoantibodies as the cause of neutropenia involved a female infant who had severe infections and was found at the age of 7 months to have a neutrophil count of 1.0 × 109/l. The peripheral blood contained fewer than 3% mature neutrophils and the bone marrow revealed virtually no mature granulocytes, although it did contain normal granulocyte precursors. The patient’s serum contained the neutrophil-specific autoantibody anti-HNA-1b with a titre of 16–256. The antibody was mainly IgG and the patient was HNA-1b positive. After steroid therapy, the granulocyte count rose to 310 × 109/l and the leucoagglutinin titre fell to 2, but the patient relapsed when steroids were discontinued (Lalezari et al. 1975). The syndrome is now well established (McCullough 1988; Bux et al. 1998).
Autoimmune neutropenia (AIN) in children is traditionally divided into two forms. In so-called ‘primary’ AIN, neutropenia is the sole abnormality and, although neutrophil counts may fall below 0.500/l, bacterial infections, when they occur, are generally mild. Primary AIN is commonly diagnosed between the ages of 5 and 15 months, but has been observed as early as day 33 of life (Bux et al. 1998). Spontaneous remission occurs in 95% of the patients within 7–24 months, but neutropenia may persist for a few years. A high percentage of autoantibodies (35–86%) bind preferentially to granulocytes from HNA-1a and HNA-1b homozygous donors, but other specificities have been found (Bux et al. 1998; Bruin et al. 1999). The bone marrow is typically normocellular or hypercellular, with a variably diminished number of segmented granulocytes. For severe infections or prior to surgery, G-CSF, corticosteroids and IVIG (19 mg/kg per day) can effect neutrophil increases of 50–100% and each has been used successfully in many cases (Pollack et al. 1982; Bussel and Lalezari 1983; Bux et al. 1998).
Secondary AIN occurs in association with other autoimmune diseases. In contrast with primary AIN, infections are usually more severe and the autoantibody is more commonly directed against FcγRIIIb (Shastri and Logue 1993; Bruin et al. 1999).
Drug-Induced Immune Granulocytopenia
Mechanisms responsible for drug-induced immune neutropenia are similar to those involving red cells (see Chapter 7). The classic case of pyramidon-induced granulocytopenia described by Moeschlin and Wagner (1952) is an example of the mechanism in which the drug does not bind firmly to the cells, but in which drug, antibody and a determinant on the cell membrane form a trimolecular complex. Although quinine is usually implicated in drug-induced thrombocytopenia, it may be involved rarely in cases of drug-induced neutropenia in which the quinine antibodies react with the same glycoprotein as anti-HNA-2a, and/or an 85-kDa glycoprotein (Stroncek et al. 1994). Drug-induced neutrophil antibodies may be directed against a metabolite of the drug (Salama et al. 1989). Recombinant G-CSF has reportedly shortened the period of granulocytopenia in some of these cases.
Reactions to Granulocyte Transfusions
Granulocyte transfusion recipients sometimes produce antibodies specific to HNA-1a, HNA-1b and HNA-2a (see also Chapters 14 and 15). Further transfusion of granulocytes to patients with these antibodies may lead to severe febrile and pulmonary transfusion reactions (Stroncek 1996). Haematopoietic progenitor cell transplant recipients who produce HNA-2a antibodies as a result of granulocyte transfusions have experienced marrow graft failure (Stroncek et al. 1993b).
Tests for Granulocyte Antibodies and Antigens
HNA antigens were initially characterized by serology using cellular assays that are labor-intensive and difficult to perform. As with the HLA system, genotyping for antigens and solid phase assays for antibody detection are gradually replacing these older tests (Stroncek and Clay 2011). Genotyping assays are now available for the HNA-1, HNA-3, HNA-4, and HNA-5 systems. A simple genotyping method is not yet available for HNA-2a. However HNA-2a is located on CD177 and CD177 monoclonal antibodies specific to the HNA-2a antigen are commercially available. MoAbs to CD16, CD11b, and CD11a, which carry HNA-1, HNA-4, and HNA-5 antigens are available as well.
The following three techniques, likely rendered obsolete by the next edition of this volume, are still in common use in reference laboratories to detect granulocyte-specific antibodies: (1) granulocyte agglutination; (2) immunofluorescence; (3) chemiluminescence; and (4) monoclonal antibody-specific immobilization of granulocyte antigen assays.
Granulocyte Agglutination Technique
Pure granulocyte suspensions are prepared by dextran sedimentation followed by centrifugation of the supernatant on Ficoll-hypaque (density 1.077). The contaminating red cells in the granulocyte pellet at the bottom of the tube are lysed with ammonium chloride or distilled water. Alternatively, granulocytes can be isolated by double-density gradient centrifugation. Agglutination techniques are carried out in microplates.
Granulocyte Immunofluorescence Technique
Purified suspensions of granulocytes are prepared as described above. The granulocytes are fixed with paraformaldehyde, incubated with the serum to be tested, then washed and finally incubated with fluorescein isothiocyanate-labelled anti-Ig serum. The Fab or F(ab′)2 fragments of the IgG fraction of anti-human Ig are used because whole IgG anti-Ig tends to bind to the Fc receptor on granulocytes. With the above modifications, the fluorescein-labelled antiglobulin test is more sensitive than granulocyte agglutination for the detection of IgG antibodies (Verheugt et al. 1977).
When flow cytometry is used for the granulocyte immunofluorescence technique (GIFT) instead of microscopy, there is no need for isolating granulocytes, as granulocytes can be identified according to light scatter patterns. Furthermore, granulocytes, platelets and lymphocytes can be tested simultaneously (Robinson et al. 1987). Flow cytometry has been found to be slightly more sensitive for the detection of granulocyte antibodies than the GIFT (Sintnicolaas et al. 1991).
Both granulocyte-specific and HLA-A, -B and -C antibodies are detected in the GIFT. The lymphocytotoxicity test (LCT) and the immunofluorescence test are more sensitive for the detection of HLA class I antibodies. If a serum is negative in these tests but positive in the GIFT, the serum is very likely to contain granulocyte-specific antibodies. If the tests on lymphocytes are positive and if it is necessary to ascertain whether granulocyte-specific antibodies are present, the serum should be absorbed with pooled platelets to remove any HLA class I antibodies or the serum must be tested with a granulocyte panel typed for granulocyte specific antigens. Unfortunately, positive reactions with lymphocytes particularly in the immunofluorescence test may be due to lymphocyte-specific antibodies, in which case it may be difficult to ascertain the presence of granulocyte-specific antibodies, unless a known specificity is detected.
With the GIFT, not only antibodies but also preformed immune complexes cause positive reactions, due to adherence to Fc and complement receptors (Camussi et al. 1979; Engelfriet et al. 1984). There are three possible ways of distinguishing between antibodies and fixed immune complexes:
Chemiluminescence Test
To prepare suspensions of mononuclear cells and granulocytes, fresh EDTA blood is centrifuged on Ficoll-hypaque (density 1.077). The mononuclear cell fraction is washed three times. The red cell/granulocyte fraction is resuspended in PBS and mixed 1 : 4 with dextran solution. After sedimentation (30 min) the granulocytes in the supernatant are packed and then washed twice in PBS. The granulocytes are resuspended in PBS and transferred to a glass tube pre-located in a waterbath at 52°C and after 2 min are allowed to cool in another tube at room temperature. The purpose of the heat treatment is to render the granulocytes incapable of responding to immunoglobulin aggregates in the sera to be tested which would lead to non-specific generation of chemiluminescence.
For the assay, granulocytes are incubated with serum and then washed in phosphate-buffered solution (PBS) and resuspended in Hanks’ BSS. The granulocytes are then incubated with freshly prepared mononuclear cells from the peripheral blood and with luminol. Chemiluminescence is generated as a result of phagocytosis of sensitized granulocytes, which leads to the formation of oxygen radicals and oxidation of luminol. Chemiluminescence is measured in a luminometer (Hadley and Holburn 1984). The sensitivity of the chemiluminescence (CL) test is similar to that of the GIFT (Lucas 1994).
Phenotyping and Genotyping of Neutrophil Antigens
Phenotyping
By tradition, neutrophil antigen typing has been performed using human alloantibodies in the granulocyte agglutination or GIFT assay. However, MoAbs are rapidly replacing these reagents. These reagents have been used to phenotype neutrophils by flow cytometry. This method is faster and easier than manual methods with alloantibodies, as whole blood rather than isolated neutrophils can be used.
Genotyping
Genotyping assays are performed with DNA isolated from whole blood, thus eliminating the need to isolate granulocytes. Furthermore, leucocyte DNA can be stored for months before testing is performed. The characterization of the genes encoding HNA-1 antigens has led to the development of assays for these antigens (Bux et al. 1995b; Hessner et al. 1996). Genotyping of HNA-1 antigens is particularly valuable given the rarity of alloantibodies to HNA-1c. Genotyping for FCGR3B alleles is complicated by the high degree of homology between FCGR3B and FCGR3A. Among the five nucleotides that differ between FCGR3A*1 and FCGR3A*2, FCGR3A is the same as FCGR3A*1 at three nucleotides and the same as FCGR3A*2 at two nucleotides. As a result, most laboratories use PCR and sequence-specific primers to distinguish FCGR3A alleles. A unique set of primers is used to amplify each of the three alleles. Genotyping is available for the HNA-1, HNA-3, HNA-4, and HNA-5 systems. Unfortunately, HNA-2a genotyping reagents are not available. The HNA-2a-negative phenotype is caused by CD177 mRNA splicing defects (Kissel et al. 2002). However, no mutations have been detected in the CD177 genomic DNA from subjects with HNA-2a-negative neutrophils. It may be possible to distinguish positive and negative phenotypes by analysing CD177 mRNA for accessory sequences, but this is a highly sophisticated methodology.
Cold Autoantibodies to Lymphocytes
Anti-I and Anti-i
See Chapter 4.
Antigens Found Only on Monocytes
In addition to HLA class I and class II antigens, and the antigens shared by monocytes and granulocytes mentioned above, monocytes carry alloantigens that do not occur on other blood cells. Some of these antigens (EM antigens) are also present on endothelial cells (Moraes and Stastny 1977; Claas et al. 1980; Cerilli et al. 1981; Stastny and Nunez 1981); others are monocyte specific (Cerilli et al. 1981; Baldwin et al. 1983; Paul 1984). Antibodies against EM antigens are detrimental to transplanted kidneys and may be involved in GvHD. EM antibodies and antibodies reacting with monocytes, tubular endothelium and kidney cells in the cortex can be eluted from rejected kidneys (Joyce et al. 1988). The significance of monocyte-specific alloantibodies needs further evaluation.
Antigens of Platelets
Antigens Shared with Other Cells
HLA Antigens
Platelets possess mRNA encoding HLA class I molecules and are capable of synthesizing them. HLA-A,-B, and -C locus antigens have been detected serologically. Most of the HLA antigen detected on platelets is integral membrane protein, whereas a small amount is adsorbed from the plasma. Expression of class I antigens on platelets varies greatly in different subjects. Class II antigens are not detectable on platelets but HLA-DR antigens can be induced at the platelet surface by stimulation with cytokines, for example gamma-interferon, both in vitro and in vivo (Boshkov et al. 1992). Only a small proportion of the A, B and C antigens on platelets are absorbed from the plasma (Santoso et al. 1993a).
Red Cell Antigens Also Found on Platelets
ABH, Lewis, I, i and P antigens on platelets are described in Chapter 4. Using a sensitive two-stage radioimmunoassay the major antigens of the Rh, Duffy, Kell, Kidd and Lutheran systems have been shown to be absent from platelets (Dunstan et al. 1984). Although expression of ABH antigens on platelets is generally insufficient to reduce the survival of ABH-incompatible platelets in transfusion recipients, anecdotal reports of poor survival of A and B mismatched platelets led to the discovery of donors who are ‘high expressers’ (Curtis et al. Blood 2000). The A and B antigen expression on platelets of 100 group A(1) and group B blood donors revealed that 7% and 4%, respectively, had platelets expressing A and B antigen levels substantially exceeding mean levels. Serum A(1)- and B-glycosyltransferase levels of A and B high expressers were significantly elevated as well. H antigen levels are reduced on the red cells of high expressers. The molecular basis for the high-expresser trait has not been elucidated.
Antigens Found Only on Platelets (Platelet-Specific Antigens)
Several systems have been defined whose antigens are found on polymorphic glycoproteins on platelet membranes. These antigens are not truly ‘platelet specific’ as some can be expressed by other cells such as endothelial cells. The human platelet antigen (HPA) nomenclature system was adopted in 1990 (von dem Borne and Décary 1990). The HPA nomenclature categorizes all alloantigens expressed on the platelet membrane, except those encoded by genes of the major histocompatibility complex. A platelet-specific alloantigen is accepted by an international nomenclature committee as an HPA when its molecular basis has been defined. The different HPAs are then grouped into systems based on serologic detection by human alloantibodies defining a given alloantigen and its ‘antithetical’ alloantigen. To date, 24 platelet-specific alloantigens have been defined by immune sera, of which 12 are grouped into six biallelic systems (HPA-1, -2, -3, -4, -5, -15) (Table 13.2 and Metcalfe et al. 2003). For the remaining 12 antibodies, alloantibodies against the antithetical antigen have yet to be discovered. The molecular basis of 22 out of the 24 serologically defined antigens has been resolved. In all but one, the difference involves a single amino acid substitution generally caused by a single nucleotide polymorphism (SNP) in the gene encoding the relevant membrane glycoprotein. The systems are numbered in the order of the date of publication and the antigens are designated alphabetically in the order of their frequency in the population. This nomenclature has been criticized, the main objection being that HPA-1a, HPA-4a, HPA-6a, HPA-7a and HPA-8a are five different names for identical GPIIIa molecules carrying these high-frequency antigens. Only the GPIIIa molecules that carry the low-frequency antigens of these systems differ from each other due to amino acid substitutions at different positions of the molecule (Newman 1994).
Table 13.2 Human platelet antigens (HPAs).
Source: Adapted from Metcalfe et al. 2003. Reproduced with permission of John Wiley & Sons Ltd.
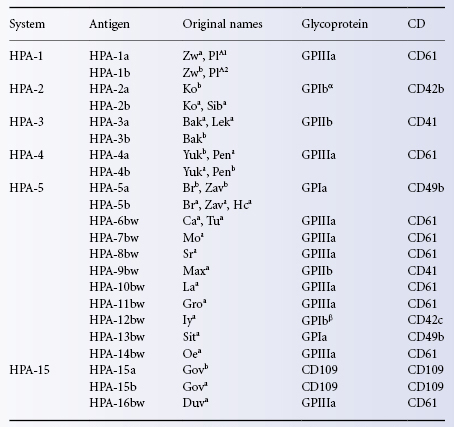
HPA-1 System (Zw, PlA)
The first system to be described was recognized by van Loghem and co-workers (1959) when a serum was found that agglutinated some samples of platelets but not others; the antigen was named Zwa when an antithetical antigen (Zwb) was recognized (van der Weerdt et al. 1962, 1963). Anti-PlA1 (Shulman et al. 1961) was subsequently shown to have the same specificity as anti-Zwa. The system is now named HPA-1 and the antigens, HPA-1a and HPA-1b. Ninety-eight per cent of white people are HPA-1(a+) and 27% HPA-1 (b+).
The HPA-1 gene has two alleles, HPA-1a and HPA-1b. Anti-HPA-1a is associated with most cases of post-transfusion purpura and neonatal alloimmune thrombocytopenia (NATP).
Related posts:
 Red cell antibodies against self-antigens, bound antigens and induced antigens
Red cell antibodies against self-antigens, bound antigens and induced antigens
 Blood grouping techniques
Blood grouping techniques
 Alternatives to allogeneic transfusion
Alternatives to allogeneic transfusion
 The transfusion of platelets, leucocytes, haematopoietic progenitor cells and plasma components
The transfusion of platelets, leucocytes, haematopoietic progenitor cells and plasma components
 The Rh blood group system (including LW and RHAG)
The Rh blood group system (including LW and RHAG)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


