Specificity activation
Non-specific (activate systemic immunity, relatively old)
Specific (target cancer cells or cancer-specific antigens, relatively old)
Active immunization (to elicit immunity in vivo)
Biological response modifier
Cancer vaccine
Dendritic cell therapy
Passive immunotherapy (to elicit immunity in vitro)
Lymphokine-activated killer cell therapy
Natural killer cell therapy
Cancer specific-antibodies
Table 13.2
Clinical trials for ovarian cancer—active immunotherapy
Therapy | Immune response | Clinical response | Report |
|---|---|---|---|
FR-specific gene-modified T-cell therapy | FR-specific IFN-γ production | No objective response | Kershaw, Clin Cancer Res, 2006; 12:6106 |
HER2-/MUC1-derived peptide sensitized DC vaccine | Specific IFN-γ-producing T cell, specific CTL activity in 2/3 | 2/3 SD | Brossart P, Blood, 2000; 96:3102 |
HER2-derived peptide vaccine | specific IFN-γ-producing T cell in 1/1 | PD | Murray, Clin Cancer Res, 2002; 8:3407 |
HER2-derived peptide vaccine | Immune response in ELISPOT in 1/2 | PD in 2/2 | Knutson, Clin Cancer Res, 2002; 8:1014 |
NY-SO-1-derived peptide vaccine | specific antibody in 10/13, peptide specific T cell in 3/5 | CR in 1 (at least) | Odunsi, Pro NAS, 2007; 104:12,837 |
NY-SO-1derived peptide vaccine | Peptide-specific T cell in 7/9 | Remission-free in 3/9 | Diefenbach, Clin Cancer Res, 2008; 14:2740 |
P53-derived peptide vaccine | Peptide-specific T cell in 20/20 | SD in 2/20 | Leffers, Int J Cancer, 2009; 125:2104 |
P53-derived peptide vaccine | Immune response in tetramer assay in 9/13 | Median OS, 40.8M | Rahma, Cancer Immunol immunother, 2012; 61:373 |
P53-derived peptide sensitized DC vaccine | Immune response in tetramer assay in 5/6 | Median OS, 29.6M | Rahma, Cancer Immunol immunother, 2012; 61:373 |
P53-derived peptide vaccine + low-dose cyclophosphamide | Immune response in ELISPOT in 9/10 | SD in 2/10 | Vermeij, Int J Cancer, 2012; 131:E670 |
Mannan-MUC1 fusion protein sensitized DC vaccine | Specific IFN-γ-producing T cell in 9/10 | SD > 10Y in 2/10 | Loveland, Clin Cancer Res, 2006; 12:869 |
MUC1-specific Th1-type CD4+ effector cell therapy | MUC1-specific CTL in all | CR in 1/4 | Dobrzanski, Cancer Immunol Immunother, 2012; 61:839 |
WT-1 peptide vaccine | Delayed cutaneous hypersensitivity in 5/6 | SD in 2/6 | Ohno, Anticancer Res, 2009; 29:4779 |
Anti-CA-125 idiotypic antibody (ACA125, abagovomab) | Specific anti-anti-idiotypic antibody in 28/42 | Significant prognostic improvement in patient with immune response | Wagner, Clin Cancer Res, 2001; 7:1154 |
Anti-CA-125 idiotypic antibody (ACA125, abagovomab) | Specific anti-anti-idiotypic antibody in 81/119 | Significant prognostic improvement in patient with immune response | Reinartz, Clin Cancer Res, 2004; 10:1580 |
Tumor cell sensitized DC vaccine | Specific IFN-γ-producing T cell in 2/6 | >SD response in 4/6 | Hernando, Cancer Immunol Immunother, 2002; 51:45 |
Table 13.3
Clinical trials for ovarian cancer—passive immunotherapy
Therapy | Case | Clinical response | Report |
|---|---|---|---|
Anti-CA-125 Ab (B43.13; oregovomab) | 32 | >SD response in six with Ab2-positive case | Baum, Cancer, 1994; 73(3 Suppl):1121 |
Anti-CA-125 Ab (B43.13; oregovomab) | 20 (recurrent) | Significant prognostic improvement in patient with immune response | Gordon, Gynecol Oncol, 2004; 94: 340 |
Anti-CA-125 Ab (B43.13; oregovomab) | 145 (post-remission) | No PFS improvement, but significant prognostic improvement in patient with immune response | Berek, J Clin Oncol, 2004; 22:3507 |
Anti-CA-125 Ab (B43.13; oregovomab) | 373 (post-remission) | No PFS improvement | Berek, J Clin Oncol, 2009; 27:418 |
Anti-CA-125 Ab (B43.13; oregovomab) | 40 (stage III/IV) | Braly, J Immunother, 2009; 32:54 | |
Anti-FrαAb (MORAb-003; farletuzumab) | 25 (recurrent, refractory) | SD in 9, CA-125 decrease in 2 | Konner, Clin Cancer Res, 2010; 16:5288 |
Anti-EpCAM x anti-CD3 Ab (catumaxomab) | 23 (with ascites) | No need of abdominocentesis in 22/23 | Burges, Clin Cancer Res, 2007; 13:3899 |
Anti-EpCAM x anti-CD3 Ab (catumaxomab) | 129 (with ascites) | Prolonged duration to next abdominocentesis | Heiss, Int J Cancer, 2010; 127:2209 |
Anti-EpCAM x anti-CD3 Ab (catumaxomab) | 45 (recurrent, refractory) | PR in 1, SD in 7 | Baumann, Gynecol Oncol, 2011; 123:27 |
13.2.1 Active Immunotherapy for Ovarian Cancer (Table 13.2)
13.2.1.1 Immunotherapy that Targets MUC1
MUC1 is highly expressed in many ovarian cancers and has been a primary target candidate for immunotherapy. Dobrzanski and colleagues conducted phase I and phase II studies by using a Th1 type of self-replenishing CD4+ T cells producing IL-10 and IFN-γ to combat recurrent ovarian cancer [8]. One of the four cases showed remission, and another showed a tumor-bearing survival of 16 weeks; however, the remaining two cases died of cancer within 3–5 months. T cells from long-term survivors showed IFN-γ production and an increase in the number of memory cells and TNF family ligands. Moreover, the therapy was likely to contribute to the survival of ovarian cancer patients by affecting the percentage of the regulatory T-cell subsets and by improving the number of memory CD4+ T cells.
13.2.1.2 Vaccine Therapy with p53 Peptide
Genetic aberrations of p53 and abnormal accumulation of p53 protein have been observed in the majority of serous ovarian cancers. In a cohort of stage III, stage IV, and recurrent ovarian cancer patients with no obvious disease, Rahma et al. compared one group directly administered with p53(264–272) peptide (group A) and another group administered with dendritic cells expressing the p53(264–272) peptide (group B) in a phase II study in the USA [10] and observed a tumor immune response in 69% of the group A patients and 83% of the group B patients. Progression-free survival (PFS) was 4.2 months and 8.7 months, respectively. Because there was no significant difference, they concluded that simple subcutaneous administration may be sufficient.
On the other hand, a Dutch research group conducted a phase II study for recurrent ovarian cancer by using a vaccine comprising a long-chain peptide of p53 (p53-synthetic long peptide, p53-SLP). Only two of the 20 cases showed stable disease, and they did not show a p53-specific immune response. The researchers concluded that there was no obvious effect of p53 peptide on improving the subsequent chemosensitivity or PFS [9, 10].
13.2.1.3 Immunotherapy Targeting HER2-Derived Peptide
Since HER2 is known to be highly expressed in many ovarian cancers, it is considered a good immunotherapy target. However, there was no significant clinical effect observed with by a vaccine using p369–p377 (Table 13.2). Although the trial or similar attempts using the DC vaccine are ongoing, a clinically useful vaccine has not been developed.
13.2.1.4 Vaccine Therapy Targeting WT-1
WT-1 is known to be expressed in more than half of serous ovarian cancers. A phase II trial use a WT-1 peptide as a vaccine against ovarian cancer has been performed in Japan. In 12 cases of treatment-resistant ovarian cancer, SD was noted in three cases, and the remaining nine cases were PD [11].
13.2.1.5 Active Immunotherapy Targeting CA-125
Because CA-125 is a specific protein that is expressed in a majority of ovarian cancers, it has been considered to be a good immunotherapy target. Aside from oregovomab (which will be discussed later), another potential immunotherapy for CA-125 includes the use of an anti-idiotypic antibody against the anti-CA-125 antibody ACA-125 (abagovomab). Since ACA-125 is structurally similar to CA-125, it was expected that abagovomab would elicit antitumor immunity against CA-125 if administered as a vaccine. Wagner et al. reported that in 42 patients with recurrent ovarian cancer, the immune response after administration of abagovomab (which was measured as the production of Ab3) was correlated with improved prognosis [12]. Reinartz et al. also reported that in 119 patients with ovarian cancer, individuals with a good immune response showed a significantly better outcome [13].
13.2.2 Passive Immunotherapy for Ovarian Cancer (Table 13.3)
13.2.2.1 Passive Immunotherapy with Anti-CA-125 Antibody
Large-scale development of the immunotherapy reagent oregovomab, a mouse monoclonal antibody B43.13 against CA-125, has been produced. The initial exploratory study showed that among the patients administered oregovomab, patients in whom anti-idiotypic antibodies (Ab2) and T-cell immunity have been induced showed a better tendency of prognosis and elicited the expected immunotherapeutic response to oregovomab (Table 13.3). Then, Berek et al. conducted a randomized phase II trial in which oregovomab was administered as a maintenance therapy to 145 patients with recurrent ovarian cancer after postoperative TC therapy. This study showed that the PFS of patients with increased Ab2 levels was 18.8 months, while that of the cases with a weak immune reaction was 6.1 months; the PFS of the placebo group was 10.3 months [14]. Unfortunately, a phase III trial with 373 cases failed to reproduce the results of the phase II study [15]. Thus, a single use of oregovomab did not show an apparent clinical effect. However, another research group indicated that based on the results of a randomized phase II study of 40 cases of ovarian cancer, the combination of paclitaxel/carboplatin chemotherapy with oregovomab may augment antitumor immunity [16].
13.2.2.2 Immunotherapy with an Antibody Against the Folate Receptor
Elevated expression of folate receptor α, which is thought to be involved in cancer growth, has been shown in ovarian cancer as well as in many other cancers. A treatment effect with anti-folate receptor antibodies against ovarian cancer has been reported in expiratory clinical trials (Table 13.3). In 2007, MORAb-003 (farletuzumab), a humanized antibody against folate receptor, was developed by Morphotek, Inc. In the phase I study for platinum-resistant ovarian cancer, 36% of patients maintained SD [17]. Furthermore, phase II trials for platinum-sensitive and platinum-resistant ovarian cancer have been reported. Currently, a phase III trial using the combination of chemotherapy and farletuzumab is underway [18].
13.2.2.3 Immunotherapy Using Antibodies Against EpCAM and CD3
Another promising passive immunotherapy for ovarian cancer targets epithelial cell surface antigen (EpCAM). Catumaxomab is a double antibody against both EpCAM and CD3. Heiss et al. examined the inhibitory effect of catumaxomab on cancerous ascites and showed that the period to next puncture was significantly extended in the administration group [19]. Furthermore, Baumann et al. reported the clinical effect of catumaxomab in ovarian cancer [20] (Table 13.3).
13.2.3 Problems Toward the Development and Clinical Application of Immunotherapy
As described above, there have been continuous attempts to develop immunotherapy for ovarian cancer, and recently, clinical efficacy has been shown in some instances. However, despite a long history of immunotherapy against solid cancers, the effect of cancer immunotherapy has been limited because there are several problems in its development. Every time new knowledge of tumor immunity was discovered in the basic fields, the new idea of immunotherapy was usually evaluated in preclinical trials with animal experiments similar to the process of evaluating other anticancer reagents. In this step, in vivo mouse models play an important role as an immunotherapy model, but there is inherent problem with the use of mouse models as the evaluation system. The use of established cancer cell lines along with pure mouse strains is thought to mimic the immune reaction of human immunity. This experimental system has been established using cancer cell lines from a wide variety of organs. The effect of the immunotherapies is validated in animal models as the preclinical phase and is eventually administered to actual cancer patients in clinical trials. However, in most cases, only a small percentage of the patients show efficacy despite a marked effect in the animal experiments. One of the reasons is that the immune reaction is a highly complex biological phenomenon compared to chemotherapy agents. In the case of chemotherapy agents, a mouse model system using transplanted tumor has much in common with human cancers, and the effectiveness of a therapy in animal experiments may predict clinical efficacy. In contrast, animal models of cancer immunotherapy are only a simplified model of the true complex tumor immunity in humans, and there is a large gap between them. For example, an established murine cell line often grows rapidly in vivo, but human cancers can maintain a state of dormancy for years. Such differences may influence the evaluation of tumor immunity.
Second, the evaluation method in clinical trials is also challenging regarding the clinical efficacy of immunotherapy. With chemotherapy agents, evaluating the tumor size may be associated with long-term efficacy. However, in case of immunotherapy, an initial antitumor effect does not necessarily correspond to the final clinical efficacy. Compared to conventional chemotherapies, immunotherapy requires a longer time interval to elicit an antitumor effect. However, we are unaware of the exact signaling mechanisms in the immune system, and we do not have a reliable method to predict the final effect of immunotherapy. Similar to other molecularly targeted drugs, it is important to develop effective predictive biomarkers in order to efficiently implement immunotherapy.
13.3 Immune Checkpoint Inhibition in Ovarian Cancer
Several years ago, a novel immunotherapy attracted a great deal of attention. A therapy using an anti-PD-1 antibody, which inhibits the binding of PD-L1 to PD-1, was used for malignant melanoma, renal cancer, and lung cancer. The first trial showed that the antibody had a high antitumor effect not only in melanoma and renal cancer (which have high immunogenicity) but also in lung cancer, which is not considered to be immunogenic [21, 22].
13.3.1 Basic Mechanism of Function of Immune Checkpoint Molecules
Generally, an immune reaction to antigens (including cancers) is initiated with antigen recognition by antigen-presenting cells (APCs) such as dendritic cells (cognitive phase). Following antigen recognition, dendritic cells migrate to lymph nodes and present specific antigens to T cells via MHC class II molecules. As a result, T cells, including CD8-positive cytotoxic T cells, recognize the existence of cancer and become activated in a tumor-specific manner via the T-cell receptor (TCR). This interaction between the MHCs on APCs and TCRs on T cells is called the “first signal.” At the same time, a “second signal” is sent via interaction of specific molecules known as immune checkpoint molecules. If this interaction occurs between B7 and CD28, active immunity is initiated. In contrast, if the interaction occurs between B7 and CTLA-4 (cytotoxic T lymphocyte-associated protein 4), there is inhibition of the immune response [23].
This type of pro-/anti-immune mechanism also exists in local immunity when T cells recognize their targets (effector phase). During this phase, the interaction between PD-1 on T cells and PD-L1 on target cells is thought to result in the attenuation of the immune response. Therefore, if tumor cells express PD-L1, there is a reduction in the immune attack by T cells [24] (Fig. 13.1).
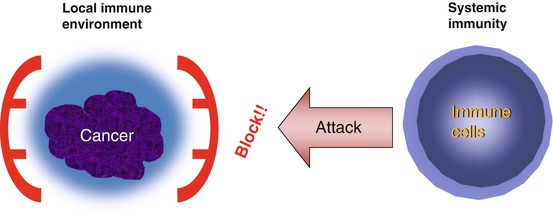
Fig. 13.1
Tumor immune escape hypothesis. This hypothesis is proposed to explain why systemic immunotherapy has not been successful. According to the hypothesis, by using unknown mechanism, e.g., expressing some immunoinhibitory molecules, tumor cells keep their local immune environment in immunosuppressive state. Therefore, even if we can successfully elicit potent systemic antitumor immunity, it does not effectively reach tumor cells
These immunoinhibitory molecules are considered to serve as cancer immune escape machinery; thus, inhibition of these signals is expected to be a target for potent immunotherapies (Fig. 13.1).
13.3.2 Immune Checkpoint Inhibition as a Cancer Immunotherapy
In 1999, clinical trials using the anti-CTLA-4 antibodies ipilimumab and tremelimumab were initiated [25, 26]. Ipilimumab is a humanized IgG1-type anti-CTLA-4 antibody and is currently used clinically to treat malignant melanoma. On the other hand, antibodies against PD-1/PD-L1 have been used in various cancers [6, 21, 22]. Nivolumab is a current treatment for malignant melanoma and lung cancer in Japan and the USA, and pembrolizumab has also been approved for use against these cancers in the USA. In addition, the anti-PD-L1 antibody atezolizumab is administered to treat urothelial cancer. At present, many immune checkpoint inhibitors are being developed and are expected to have clinical applications in the near future.
13.3.3 Rationale of Immune Checkpoint Inhibition in Ovarian Cancer
Failure of conventional cancer immunotherapies has brought forth the idea of an immune escape mechanism in tumors. According to this theory, cancer cells actively alter and attenuate their local micro-immune environment by expressing immunosuppressive molecules (Fig. 13.2a). Thus, simply strengthening the immunity of the whole body is insufficient to achieve a therapeutic effect due to the local immune escape mechanism (Fig. 13.1). We and other researchers have shown that the local immune environment, especially tumor infiltration of CD8+ T cells, is closely associated with outcome of patients with ovarian cancer [27–29]. Furthermore, we showed that PD-L1 expression in ovarian cancer is a prognostic factor for ovarian cancer, and its expression is negatively associated with CD8+ T-cell infiltration, suggesting that the expression of PD-L1 in tumor cells plays a major role in the suppression of local immunity [29].

 Ovarian Cancer Genome and Molecular Experimental Sciences
Strategies for the Management of Epithelial Ovarian Cancer
Ovarian Cancer Genome and Molecular Experimental Sciences
Strategies for the Management of Epithelial Ovarian Cancer
 Management of Ovarian Cancer in the Elderly Population
Management of Ovarian Cancer in the Elderly Population
 Strategies for the Management of Non-epithelial Ovarian Tumors
Strategies for the Management of Non-epithelial Ovarian Tumors
 Pathology of Epithelial Ovarian Tumors
Pathology of Epithelial Ovarian Tumors
 Pathology of Non-epithelial Ovarian Tumors
Pathology of Non-epithelial Ovarian Tumors




Related posts:
Ovarian Cancer Genome and Molecular Experimental Sciences
Strategies for the Management of Epithelial Ovarian Cancer
Management of Ovarian Cancer in the Elderly Population
Strategies for the Management of Non-epithelial Ovarian Tumors
Pathology of Epithelial Ovarian Tumors
Pathology of Non-epithelial Ovarian Tumors
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
