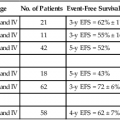
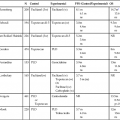
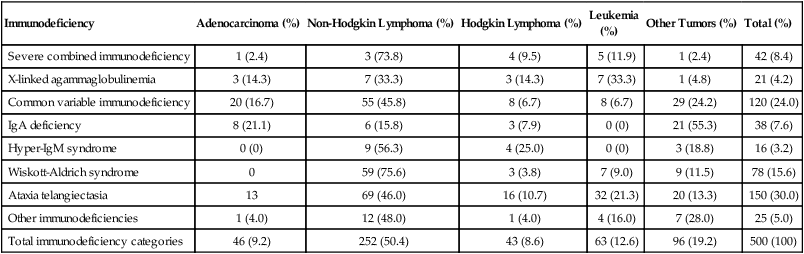
14 • Cancer remains a major cause of mortality among patients with primary and acquired immunodeficiencies. • Cancer in immunocompromised hosts is frequently associated with infectious agents, including the following: • Categories of genetic immunodeficiencies with increased risk of the development of cancer include the following: • Cancer can occur de novo, preexisting cancer can recur, or cancer can be transmitted by a donor. • In general, the outcome for immunocompromised patients with cancer is inferior to the outcome for the general cancer population. In 1959, Lewis Thomas proposed the concept that immune surveillance was an active process controlling the emergence of malignant clones from somatic cells that undergo precancerous mutations during the lifetime of a normal, immune-competent individual.1 This hypothesis predicted that immune-deficient subjects should experience much higher rates of all types of cancers compared with the general population. Indeed, data from many databases have demonstrated an increased incidence of cancer but have not substantiated an increased risk of all cancer types.2–11 In many cases, de novo, reactivated, or chronic infections play a substantial role in tumor development. Epstein-Barr virus (EBV) has been associated with B-cell lymphoproliferative disease, Hodgkin lymphoma, even leiomyosarcoma.12,13 Human immunodeficiency virus (HIV)-infected patients and solid-organ allograft recipients run an increased risk of Kaposi sarcoma, which is associated with human herpesvirus 8 (HHV8), also known as Kaposi sarcoma–associated herpesvirus.14 Additionally, HIV-infected patients and solid-organ allograft recipients are at greater risk of the development of squamous cell carcinoma of the skin, cervix, and anus associated with human papillomavirus (HPV).15–17 Immunocompromised patients, especially those with abnormalities in humoral immunity, experience higher rates of gastric carcinomas and gastric mucosa–associated lymphoid tissue (MALT) lymphomas associated with Helicobacter pylori.18,19 Even among classic primary immunodeficiencies, the array of cancers varies among the specific immunodeficiency diagnoses.2,3 The delineation of precise genetic causes for many of the primary immune deficiencies has made it possible to begin to identify specific molecular pathways to explain the differences in tumors seen among patients with some of these disorders. Patients with inherited defects of genomic instability and DNA repair, which can lead to both immune deficiency and propensity to tumor development, are at increased risk of tumors of hematopoietic and epithelial origin.4,20 Advances in prevention and treatment of opportunistic infections now allow patients with primary immunodeficiencies to enjoy longer lives. However, neoplastic disorders—particularly lymphoproliferative complications—remain a major cause of premature mortality, exceeded only by infections.2 The incidence of cancer in patients with primary immunodeficiencies increases with advancing age.2,21 Table 14-1 summarizes the types of cancers observed in patients with primary immunodeficiencies. These data came from the international Immunodeficiency Cancer Registry, which evolved in the 1970s as an outgrowth of the early clinical observations of Gatti and Good3 in immunodeficient children. A retrospective review of clinical and pathological materials from the early cases reveals some imperfections in the original cataloging. For example, it is likely that a significant proportion of the males diagnosed as having hypogammaglobulinemia and in whom lymphomas developed were actually affected with X-linked severe combined immunodeficiency (SCID). Similarly, a review of slides from cases of “leukemia” in patients with SCID, hypogammaglobulinemia, and Wiskott-Aldrich syndrome (WAS) suggest that these were more likely manifestations of non-Hodgkin lymphoma (NHL). Nonetheless, the general outline of tumor types and their proportional distribution among patients with various immunodeficiencies remain relevant even today. Table 14-1 Immunodeficiency Cancer Registry Cases: Distribution of Tumors and Immunodeficiencies Modified from Filipovich AH, Heinitz KJ, Robison LL, Frizzera G. The immunodeficiency cancer registry: a research resource. Am J Pediatr Hematol Oncol 1987;9:183–4. The vast majority of cancer in persons with primary immunodeficiencies are lymphoid in origin and are associated with EBV. EBV has been associated not only with B-cell malignancies but also with T-cell malignancies, Hodgkin lymphoma, and gastric carcinoma.12 However, other infectious agents are associated with cancers observed in patients with primary immunodeficiencies, such as H. pylori in MALT lymphomas in patients with humoral deficiencies, particularly IgA deficiency.18 The exception to this rule is cancers that arise in patients with an underlying defect in chromosomal repair, such as ataxia telangiectasia, Nijmegen breakage syndrome (NBS), or Bloom syndrome. In persons with these disorders, the cancers are usually not associated with infectious agents. The lack of immunosurveillance is less likely to be the primary defect leading to cancer development. Instead, the inability to correct genetic alterations is more likely the etiology for the increased risk of cancer in these patients. The following section provides a brief description of the known molecular defects in specific primary immunodeficiencies and the most common cancers observed. SCID is a collection of more than a dozen genetically distinct disorders with severe impairment of both cellular and humoral immune function, leading to early mortality from opportunistic infections, usually during infancy.22 EBV-associated lymphomas are almost exclusively seen in patients with SCID. However, the only SCID phenotypes at risk are those in which both B cells (targets for EBV transformation) and severe quantitative or qualitative defects in T cells are present. Examples of such types of SCID include mutations in the following: X-linked common γ-chain gene of multiple interleukin (IL) receptors (IL-2RG), JAK3, IL-7 alpha-chain, CD3 delta and/or epsilon chains, and CD45.22 EBV-associated lymphomas are seen to a lesser extent in patients with purine nucleoside phosphorylase and adenosine deaminase deficiency. In these two diseases, T-cell expansion and function are impaired by accumulation of toxic intracellular metabolites; although B cells are affected to a lesser extent, absence of B cells is common. EBV-associated lymphomas are seen to a lesser extent in persons with Omenn syndrome, caused by mutations in RAG1 genes predominantly, resulting in severe restrictions on both B- and T-cell repertoire development.22 WAS, an X-linked disorder of variable immunodeficiency and microthrombocytopenia, results from mutations in the WAS gene.23 The WAS gene encodes a large intracellular protein with several functional domains involved with cytoskeletal integrity and signal transduction. Several molecules reported to be associated with WAS are involved in normal progression through the cell cycle. WAS is expressed in cells of hematopoietic origin and in the thymus. Experimental evidence suggests that B cells in patients with WAS are resistant to apoptosis, and reports of EBV-negative B-cell lymphomas do exist, especially among men with clinically milder forms of WAS, which are sometimes termed “X-linked thrombocytopenia.” X-linked lymphoproliferative (XLP) syndrome, a condition associated with severe or fatal complications of EBV infection and a high risk of lymphoma, results from mutations in the SH2D1A or SAP gene on the X chromosome.24,25 Clinical features of XLP syndrome include an intense immune reaction to EBV associated with hemophagocytosis and liver failure, lymphomas, aplastic anemia, and/or acquired hypogammaglobulinemia.26 Slam-associated protein, an adaptor protein linked to at least four known regulatory molecules, can alter T and natural-killer cell functions in both activating and downregulating directions and is thought to be involved in T-cell/B-cell interactions through cytokine regulation.24 Contrary to early reports, it is now known that many of the lymphomas occurring in patients with XLP syndrome are EBV negative.26 A syndrome caused by a deficiency in the XIAP gene has recently been described.27 Patients have defects in their antiapoptic functions, and the disorder has been named “deficiency of X-linked inhibitor of apoptosis” (XIAP). Because patients with deficiency of XIAP frequently have problems with EBV infections, XIAP has also been called XLP2. However, patients with deficiency of XIAP do not have an increased incidence of lymphoma.28 X-linked CD40 ligand deficiency, also known as hyper-immunoglobulin M syndrome, results in failure of immunoglobulin switching by B cells (which requires signaling through CD40) and decreased development and maintenance of type 1 cell–mediated responses (including natural killer cell function) because of impaired responsiveness of CD40-expressing monocyte-derived antigen-presenting cells.29 Interestingly, patients with CD40 ligand deficiency seem to have an increased risk of EBV-associated Hodgkin lymphoma, but not NHL. Patients with CD40 ligand deficiency are also at increased risk for biliary carcinomas, because a high rate of sclerosing cholangitis occurs in patients with a history of chronic cryptosporidiosis.30 Autoimmune lymphoproliferative syndrome (ALPS) represents a constellation of genetic apoptosis defects associated with mutations in FAS, Fas ligand, and caspase 8 genes.31 Patients with ALPS usually have an increase of circulating CD3+, αβ T-cell receptor−, CD4−CD8− T cells (so-called double-negative T cells). Characteristic clinical features of ALPS include chronic multifocal lymphadenopathy, splenomegaly, and/or autoimmune cytopenias.31 Most patients experience symptomatic improvement with immunosuppressive therapy, and autoimmune complications generally lessen in severity with advancing age. The incidence of lymphoma, B-cell, T-cell, or Hodgkin lymphoma in patients with ALPS is as high as 30%, and in some patients, more than one lymphoid tumor have developed over time.32 Ataxia telangiectasia (AT) is an autosomal-recessive disorder characterized by a mutation in the ataxia telangiectasia (ATM) gene, which acts as a sensor of double-stranded DNA breakage and activates numerous damage repair pathways, including cell-cycle checkpoint control, p53 activation, and DNA repair.33,34 Mutations in ATM lead to accelerated telomere loss and premature aging.35 In the context of normal lymphopoiesis, patients with AT demonstrate a 25-fold increase in nonrandom rearrangements of immunoglobulin and T-cell receptor genes compared with lymphocytes from persons without AT.36 Thymic output in persons with AT is very reduced, and a restricted T-cell repertoire emerges from oligoclonal postthymic expansion.37 Lymphoid cancers (both lymphomas and leukemias, which are usually EBV negative and can be either T cell or B cell in origin) are seen in patients with AT. However, epithelial cancers involving the skin, gastrointestinal tract, genitourinary tract, and central nervous system also can develop in persons with AT.4 Multiple tumors can be present simultaneously but more commonly develop sequentially.3 NBS is another rare autosomal-recessive syndrome that, like AT, is associated with both humoral and T-cell defects, clinical radiosensitivity, chromosomal instability, and predisposition to lymphoid and epithelial cancers.38 Like ATM, the protein that is defective in persons with NBS (NBS1, nibrin, or p95) functions to “sense” DNA double-strand breaks and activates a diversity of corrective actions.
Immunodeficiency and Cancer
 Epstein-Barr virus (associated with lymphoproliferative disorders and leiomyosarcoma)
Epstein-Barr virus (associated with lymphoproliferative disorders and leiomyosarcoma)
 Human papillomavirus (associated with skin anal and cervical carcinomas)
Human papillomavirus (associated with skin anal and cervical carcinomas)
 Helicobacter pylori (associated with gastric carcinomas and mucosa-associated lymphoid tissue lymphomas)
Helicobacter pylori (associated with gastric carcinomas and mucosa-associated lymphoid tissue lymphomas)
 Combined defects with T-cell dysfunction (e.g., severe combined immunodeficiency or Wiskott-Aldrich syndrome)
Combined defects with T-cell dysfunction (e.g., severe combined immunodeficiency or Wiskott-Aldrich syndrome)
 Defects that inhibit lymphoid apoptosis (e.g., autoimmune lymphoproliferative syndrome)
Defects that inhibit lymphoid apoptosis (e.g., autoimmune lymphoproliferative syndrome)
 Defects of genomic instability (e.g., ataxia telangiectasia)
Defects of genomic instability (e.g., ataxia telangiectasia)
 Acquired deficiencies in T-cell immunity with increased risk of developing cancer, such as:
Acquired deficiencies in T-cell immunity with increased risk of developing cancer, such as:
 Human immunodeficiency virus infection
Human immunodeficiency virus infection
 Immunosuppression to treat autoimmune disorders
Immunosuppression to treat autoimmune disorders
 Immunosuppression after solid-organ transplantation
Immunosuppression after solid-organ transplantation
 The outcome is inferior because of the increased risk of infection and/or organ toxicity.
The outcome is inferior because of the increased risk of infection and/or organ toxicity.
Introduction
Cancer in Primary Immunodeficiencies
Pathogenesis of Cancer in Primary Immunodeficiencies
Immunodeficiency
Adenocarcinoma (%)
Non-Hodgkin Lymphoma (%)
Hodgkin Lymphoma (%)
Leukemia
(%)
Other Tumors (%)
Total (%)
Severe combined immunodeficiency
1 (2.4)
3 (73.8)
4 (9.5)
5 (11.9)
1 (2.4)
42 (8.4)
X-linked agammaglobulinemia
3 (14.3)
7 (33.3)
3 (14.3)
7 (33.3)
1 (4.8)
21 (4.2)
Common variable immunodeficiency
20 (16.7)
55 (45.8)
8 (6.7)
8 (6.7)
29 (24.2)
120 (24.0)
IgA deficiency
8 (21.1)
6 (15.8)
3 (7.9)
0 (0)
21 (55.3)
38 (7.6)
Hyper-IgM syndrome
0 (0)
9 (56.3)
4 (25.0)
0 (0)
3 (18.8)
16 (3.2)
Wiskott-Aldrich syndrome
0
59 (75.6)
3 (3.8)
7 (9.0)
9 (11.5)
78 (15.6)
Ataxia telangiectasia
13
69 (46.0)
16 (10.7)
32 (21.3)
20 (13.3)
150 (30.0)
Other immunodeficiencies
1 (4.0)
12 (48.0)
1 (4.0)
4 (16.0)
7 (28.0)
25 (5.0)
Total immunodeficiency categories
46 (9.2)
252 (50.4)
43 (8.6)
63 (12.6)
96 (19.2)
500 (100)
Severe Combined Immunodeficiencies
Wiskott-Aldrich Syndrome
X-Linked Lymphoproliferative Syndrome
X-Linked CD40 Ligand Deficiency
Autoimmune Lymphoproliferative Syndrome
Cancer in Immunodeficiency-Associated Genetic Disorders of DNA Repair
Ataxia Telangiectasia
Nijmegen Breakage Syndrome
Immunodeficiency and Cancer