- 3.1 Introduction
- 3.2 Primary antibody deficiencies [PADs]
- 3.2.1 Diagnosis of primary antibody deficiencies
- 3.2.2 Types of primary antibody deficiency
- 3.2.3 Differential diagnosis
- 3.2.4 Complications of antibody deficiencies
- 3.2.5 Management of antibody deficiencies
- 3.2.1 Diagnosis of primary antibody deficiencies
- 3.3 Combined primary T- and B-cell immunodeficiencies
- 3.3.1 Types of defects
- 3.3.2 Management of defects in cellular immunity
- 3.3.1 Types of defects
- 3.4 Primary defects in non-specific immunity
- 3.4.1 Defects in monocytes and dendritic cells
- 3.4.2 Defects in neutrophil function
- 3.4.3 Defects of effector functions of macrophages
- 3.4.4 Complex innate disorders
- 3.4.5 Complement deficiency
- 3.4.1 Defects in monocytes and dendritic cells
- 3.5 Secondary immunodeficiencies
- 3.5.1 Secondary causes of immunodeficiency
- 3.5.2 Acquired immune deficiency syndrome
- 3.5.3 Transmission of human immunodeficiency virus infection
- 3.5.4 The clinical spectrum of human immunodeficiency virus infection
- 3.5.5 Immunopathogenesis of acquired immune deficiency syndrome
- 3.5.6 Diagnosis of human immunodeficiency virus infection
- 3.5.7 Therapeutic options in acquired immune deficiency syndrome
- 3.5.8 Infections in the immunosuppressed host
- 3.5.1 Secondary causes of immunodeficiency
 Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
3.1 Introduction
Once a newborn infant leaves the sterile intrauterine environment, he or she meets many microorganisms and becomes colonized with ‘healthy bacteria’. Most microflora are non-pathogenic, so this colonization does not cause symptoms. When exposed to a pathogen that the child has not met before, clinical infection may result, expanding the child’s immunological memory and producing long-lasting immunity.
In any encounter with a microorganism, the development of infection depends on the resistance of the host balanced against the virulence of the microorganism and the size of the inoculum. Infections with certain organisms, for example Pneumocystis jiroveci, infections are unknown except in patients with underlying immunodeficiency, and such bugs are therefore known as ‘opportunistic’. Host defence factors are very variable; increased infection susceptibility can be inherited or acquired, including environmental, dietary or drug induced. Some infective agents, for example HIV or cytomegalovirus, have potent immunosuppressive effects and can cause serious disease. These causes of secondary immune deficiencies are discussed in this chapter as well as those of primary immune deficiencies.
Underlying immunodeficiency should be suspected in every patient, irrespective of age, who has recurrent, persistent, severe or unusual infections. Defects in immunity can be classified into primary disorders due to an intrinsic defect in the immune system that may be congenital or late onset, or those secondary to a known condition (Fig. 3.1). They may involve adaptive or innate immune mechanisms and maybe be permanent (genetic) or transient (if due to a viral infection). Furthermore, many defects are subtle and defy classification at present.
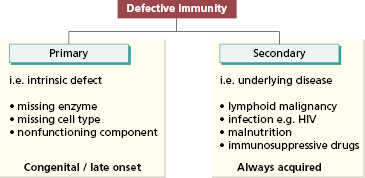
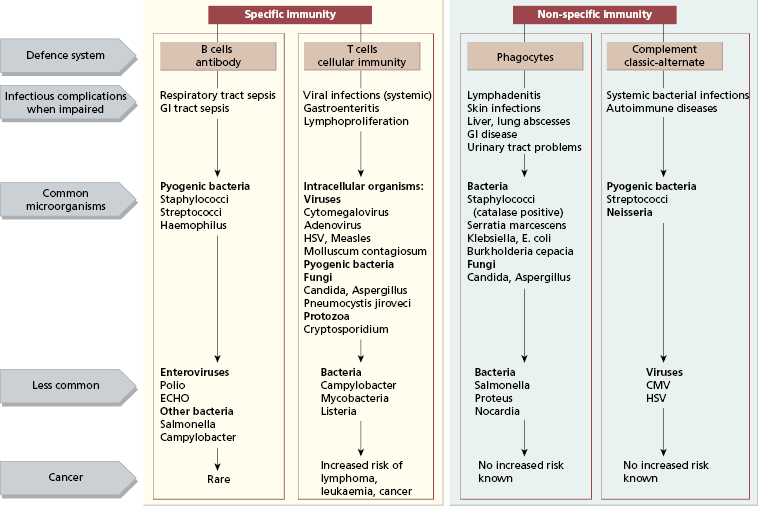
The type of organism causing the infections may give a clue to the nature of the defect (Fig. 3.2). The speed of the infection is also important. The innate immune system is the first line of defence and, if this fails, an infection will be acute and severe (even overwhelming). Infections due to defects in adaptive immunity are usually identified and treated as they often develop more slowly; in the main, bacterial infections indicate humoral (antibody and/or complement) or phagocytic defects and viral or fungal infections suggest T-cell defects.
Fig. 3.2 Defects in immunity suggested by infections with certain organisms. GI, gastrointestinal; HSV, herpes simplex virus.
3.2 Primary antibody deficiencies [PADs]
3.2.1 Diagnosis of primary antibody deficiencies
The commonest forms of primary immune deficiencies are those due to antibody failure and are discussed first (Table 3.1). These occur in children and adults and may be congenital or late onset. Congenital antibody deficiencies are rarer than late onset; >90% of patients who fail to produce protective antibodies present after 10 years of age (Table 3.2). It is easier, however, to discuss the various forms of antibody deficiencies in relation to the development of B cells, and so the clinical types are given here in this order too [Case 3.1 (lack of B cell differentiation) to Case 3.4 (failure of T cells to help B cells to immunoglobulin class switch)]. Regardless of the precise defect, the outcome is failure of production of protective antibodies resulting in increased susceptibility to bacterial infections.
Table 3.1 Prevalence of primary antibody deficiencies
| Comparison with other diseases | Per 105 population |
|---|---|
| Rheumatoid arthritis | 1000 |
| Insulin dependent diabetes | 200 |
| Multiple sclerosis | 60 |
| Systemic lupus erythematosus | 50 |
| Primary antibody deficiencies | 4–6 |
| Scleroderma | 1 |
Table 3.2 Major causes of primary antibody deficiencies in children and adults
| Age (years) | Children | Adults |
|---|---|---|
| <4 | Transient hypogammaglobulinaemia of infancy | |
| X-linked agammaglobulinaemia (XLA) | XLA (late presentation is unusual but does occur) | |
| Hyper-IgM syndromes | ||
| 4–15 | Common variable immunodeficiency disorders | |
| Hyper-IgM syndromes | ||
| Selective IgA deficiency | ||
| Selective/partial antibody deficiencies | ||
| 16–60 | Common variable immunodeficiency disorders | |
| Selective/partial antibody deficiencies | ||
| Selective IgA deficiency | ||
| Antibody deficiency with thymoma |
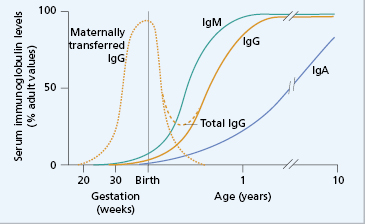
In genetic forms of antibody deficiency, recurrent infections usually begin between 4 months and 2 years of age, since maternally transferred IgG affords some passive protection for the first 3–4 months of life (Fig. 3.3). Some forms of primary antibody deficiency are inherited as X-linked or autosomal recessive traits: a history of affected relatives, especially boys, is therefore of diagnostic value, although a negative family history does not exclude an inherited condition or a de novo mutation. Primary immune deficiencies are relatively rare. A detailed history (Box 3.1) helps to distinguish them from more common causes of recurrent infection: for example, cystic fibrosis or inhaled foreign bodies are more likely causes of recurrent chest infections in childhood. However, if tests for cystic fibrosis are done, immunoglobulin measurements should always be performed at the same time.
Fig. 3.3 Serum immunoglobulin levels and age. Maternally transferred IgG (· · ·) has mostly disappeared by 6 months. As the neonate synthesizes (—) IgG, the level slowly rises, but a physiological ‘trough’ of serum IgG is characteristically seen between 3 and 6 months (- – -).
- History of repeated ENT surgery
- Lobectomy in childhood or adolescence
- Early bronchiectasis
- Skin sepsis (boils, abscesses)
- Gut infections
- Meningitis
- Streptococcus pneumoniae
- Haemophilus influenzae
- Immune thrombocytopenic purpura
- ‘Sarcoid-like’ granuloma
Clues from the examination are few: there are rarely any diagnostic physical signs of antibody deficiency, although examination often shows evidence of the consequences of previous severe infections, such as a ruptured tympanic membrane, grommets, bronchiectasis or failure to thrive.
Laboratory investigations are essential to the diagnosis. Measurements of serum immunoglobulin levels will often but not always reveal gross quantitative abnormalities. Complete absence of immunoglobulin, i.e. agammaglobulinaemia, is unusual, and even severely affected patients have very low but detectable IgG and IgM. Defects in antibody synthesis can involve one immunoglobulin isotype alone, such as IgA, groups of isotypes, often IgA and IgG, or all three major isotypes, i.e. panhypogammaglobulinaemia. The ability of a patient to make antibodies is a better guide to susceptibility to infection than total immunoglobulin levels. Failure to make specific antibody after immunization or documented exposure is fundamental to the diagnosis. Tests of specific functional antibodies are shown in Table 3.3. Measurements of IgG subclasses are not useful unless backed up by test immunizations and detection of specific IgG responses.
Table 3.3 Tests for functional antibodies (with examples in brackets)
| Detection after natural exposure/infection (chicken pox) |
| Response to prior or test immunization: |
|
|
| Caution |
| Live vaccines (e.g. BCG, MMR) should never be given to children in whom an immunodeficiency is suspected |
| Normal children under the age of 2 years do not respond to carbohydrate antigens |
Circulating B cells are identified by monoclonal antibodies to B-cell antigens (see Chapter 19). In normal blood, B cells constitute about 5–15% of the total lymphocyte population. The absence of mature B cells in an antibody-deficient individual distinguishes failed B cell differentiation, such as X-linked agammaglobulinaemia, from other causes of primary antibody deficiency in which non-functional B cells are present. Mutation analysis is essential to confirm a diagnosis of an inherited condition and enables family members to be tested and counselled. Management with replacement immunoglobulin therapy by a clinical immunologist is important (see later in the chapter).
3.2.2 Types of primary antibody deficiency (see Table 3.2 and Box 3.2)
- Common variable immunodeficiency disorders
- X-linked agammaglobulinaemia
- Hyper IgM syndromes (e.g. CD40 ligand deficiency)
- IgA and IgG subclass deficiencies
- Selective IgA deficiency
- Specific antibody deficiencies
- Transient hypogammaglobulinaemia of infancy
Transient hypogammaglobulinaemia of infancy
Maternal IgG is actively transported across the placenta to the fetal circulation from the fourth month of gestational life, although maximum transfer takes place during the final 2 months (see section 18.3.2). At birth, the infant has a serum IgG at least equal to that of the mother (see Fig. 3.3), but catabolism of maternal IgG outstrips IgG synthesized by the newborn child, resulting in a phase of ‘physiological IgG trough’. However, the normal infant is not unduly susceptible to infection because functioning antibody can be made and T cells can be activated (see section 18.3.2).
The trough in IgG is more severe if the child is premature, as IgG acquired from the mother is reduced (see Case 18.2). Improved neonatal care results in more surviving infants born between 26 and 32 weeks of gestation who are at risk of bacterial infections due to reduction of time for placental transfer. However the incidence of such infections is low in the UK where routine invasive support (e.g. indwelling lines for nutrition, monitoring, etc.) is not used. Low-birthweight babies in countries where such procedures, and the associated severe bacterial infections, are common may benefit from replacement immunoglobulin until they can synthesize their own protective antibodies (see Fig. 3.3).
Transient hypogammaglobulinaemia occurs when the otherwise normal infant is slow to start to synthesize IgG. As maternally acquired antibodies fall, the infant becomes susceptible to recurrent pyogenic infections, sometimes for many months, until spontaneous IgG synthesis begins. It is important to distinguish this condition from pathological causes of hypogammaglobulinaemia, because management differs. In most cases, the infant remains well and needs no specific therapy even though immunoglobulin levels remain below the normal range. If infections are severe, then prophylactic antibiotics should prevent further morbidity; these may be needed for 1–2 years or until endogenous IgG synthesis is satisfactory and full immunization responses documented.
X-linked agammaglobulinaemia (Bruton’s disease)
Boys with X-linked agammaglobulinaemia (XLA) usually present with recurrent pyogenic infections between the ages of 4 months and 2 years (as in Case 3.1). The sites of infection and the organisms involved are similar to other types of antibody deficiency (Figs 3.2 and 3.4), although these young boys are also susceptible to life-threatening enteroviruses.
 Case 3.1 X-linked agammaglobulinaemia (Bruton’s disease)
Case 3.1 X-linked agammaglobulinaemia (Bruton’s disease)Peter was born after an uneventful pregnancy, weighing 3.1 kg. At 3 months, he developed otitis media; at the ages of 5 months and 11 months, he was admitted to hospital with untypable Haemophilus influenzae pneumonia. These infections responded promptly to appropriate antibiotics on each occasion. He is the fourth child of unrelated parents: his three sisters showed no predisposition to infection.
Examination at the age of 18 months showed a pale, thin child whose height and weight were below the third centile. There were no other abnormal features. He had been fully immunized as an infant (at 2, 3 and 4 months) with tetanus and diphtheria toxoids, acellular pertussis, Hib and Mening. C conjugate vaccines and polio (Salk). In addition, he had received measles, mumps and rubella vaccine at 15 months. All immunizations were uneventful.
Immunological investigations (Table 3.4) into the cause of his recurrent infections showed severe reduction in all three classes of serum immunoglobulins and no specific antibody production. Although there was no family history of agammaglobulinaemia, the lack of mature B lymphocytes in his peripheral blood suggested a failure of B-cell differentiation and strongly supported a diagnosis of infantile X-linked agammaglobulinaemia (Bruton’s disease). This was confirmed by detection of a disease-causing mutation in the Btk gene. The antibody deficiency was treated by 2-weekly intravenous infusions of human normal IgG in a dose of 400 mg/kg body weight/month. Over the following 7 years, his health steadily improved, weight and height are now on the 30th centile, and he has had only one episode of otitis media in the last 4 years. He is now 12 years and able to treat himself with the same dose of subcutaneous replacement immunoglobulin at home.
Table 3.4 Immunological investigations* in Case 3.1. XLA
| Quantitative serum immunoglobulins (g/L): | ||
| IgG | 0.17 | (5.5–10.0) |
| IgA | Not detected | (0.3–0.8) |
| IgM | 0.07 | (0.4–1.8) |
| Antibody activity | ||
| Immunization responses – no detectable IgG antibodies to: | ||
| Tetanus toxoid (post Imx) | ||
| Haemophilus type b polysaccharides (post Imx) | ||
| Polio (post Imx) | ||
| Measles (post Imx) | ||
| Rubella (post Imx) | ||
| Isohaemagglutinins (IgM) not detected (blood group A Rh+) | ||
| Blood lymphocyte subpopulations (×109/L): | ||
| Total lymphocyte count | 3.5 | NR* (2.5–5.0) |
| T lymphocytes (CD3) | 3.02 | NR (1.5–3.0) |
| B lymphocytes (CD19) | <0.1 | NR (0.3–1.0) |
*Normal range for age 18 months shown in parentheses. Imx = immunisation
In almost all patients, circulating mature B cells are absent but T cells are normal. There are no plasma cells in the bone marrow, lymph nodes or gastrointestinal tract. Differentiation of pre-B cells into B cells depends on a tyrosine kinase enzyme – known as Bruton’s tyrosine kinase (Btk), normally found in developing B cells, but not mature B cells (Fig. 3.5). This enzyme, like the T-cell counterpart, Itk, interacts with lipids on the inner cell surface membrane clustered around an antigen receptor and is crucial for maturation. The Btk gene is mutated in XLA patients, resulting in either failure to synthesize the Btk protein or an abnormal enzyme that is always non-functional.
Fig. 3.5 An overview of the steps in B-cell maturation. The proteins in red are those in which gene defects have been shown to cause specific failure of B-cell differentiation and therefore primary antibody failure.
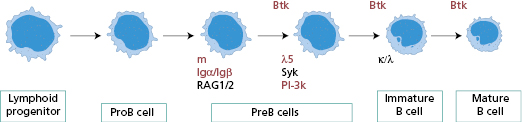
The diagnosis of XLA rests on the very low serum levels of all isotypes of immunoglobulin, the absence of circulating mature B lymphocytes and a mutation in the Btk gene. The identification of the genetic defect allows asymptomatic female carriers to be identified and counselled, and prenatal diagnosis is now feasible. The gene for Btk is located on the long arm of the X chromosome, resulting in X-linked inheritance. Other defects in the B-cell maturation pathway, though still rare, have autosomal recessive inheritance and occur in girls and boys.
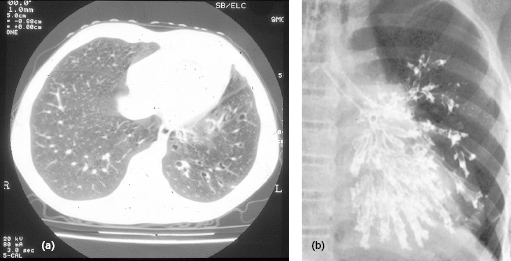
Management consists of high levels of replacement immunoglobulin for all affected individuals (see section 3.2.5), to prevent bronchiectasis (Fig. 3.6) and all types of bacterial infections.
Fig. 3.6 Bronchiectasis – (a) computed tomography scan and (b) bronchogram (no longer performed) show the typical features of damaged terminal bronchioles, leading to structural lung damage.
Hyper-IgM syndromes
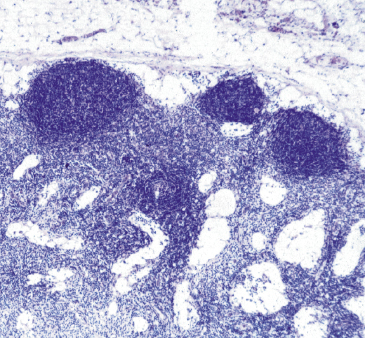
Some children with severe antibody deficiency (including boys with or without affected male relatives) have normal numbers of B cells and normal or raised serum IgM levels at presentation; such syndromes are therefore known as hyper-IgM syndromes as in Case 3.2. In the X-linked form, the boys have an additional susceptibility to Pneumocystis jiroveci infection. Such infection is normally associated with T-cell defects such as HIV or severe combined immune deficiency (see Fig. 3.2). Unlike XLA, this defect is not restricted to B cells, being due to failure of the CD40 ligand accessory molecule on T cells (Fig. 1.26). Normally, this ligand reacts with CD40 on B cells to trigger switching in specific antibody production from IgM to IgG or IgA and the formation of germinal centres. Failure of expression or functional activity of this ligand results in failure absence of switching and poor organization of the germinal centres (Fig. 3.7) and is associated with lack of memory B cells, reduced somatic hypermutation and impaired dendritic cell function to prime naive T cells. The lack of cross-linking of CD40 also results in failure of the B cells to upregulate CD80 and CD86, important co-stimulatory molecules that interact with CD28 and CTLA-4 on T cells for immune regulation and recognition of tumour cells. This accounts for the development of lymphoid and other malignancies in older patients. The lack of CD40 ligand in the thymus results in defective purging of autoreactive thymocytes, hence increased susceptibility to autoimmune diseases including neutropenia (as in Case 3.2).
 Case 3.2 CD40 ligand deficiency
Case 3.2 CD40 ligand deficiencyMichael was seen in OPD at the age of 4 years with a history of painful mouth ulcers, abdominal pain over 7 weeks but persistent diarrhoea in the last 2 weeks. He had suffered multiple episodes of ear and chest infections, starting with pneumonia at the age of 9 months, when he had been noted to have neutropenia but this had appeared to be transient. He has three healthy sisters. On examination he had multiple oral ulcers, enlarged tonsils, purulent nasal discharge, scarred tympanic membranes, abdominal distension and hepatomegaly.
He was investigated for an early presentation of inflammatory bowel disease, including stool microscopy. In addition, liver function tests and hepatitis serology were done to determine the cause of the enlarged liver, T-lymphocyte enumeration to exclude severe combined immunodeficiency (SCID) and immunoglobulin levels to exclude a CVID. Cryptosporidia were found in the stools and liver enzyme levels were raised. Serum IgG and IgA levels were very low but B and T-cell numbers were normal (see Table 3.5). Since C-reactive protein (CRP) and albumin serum levels were normal, an intestinal biopsy was not indicated. A diagnosis of primary antibody deficiency with cryptosporidiosis was made.. Abdominal ultrasound showed a diffusely enlarged liver with a dilated common bile duct.
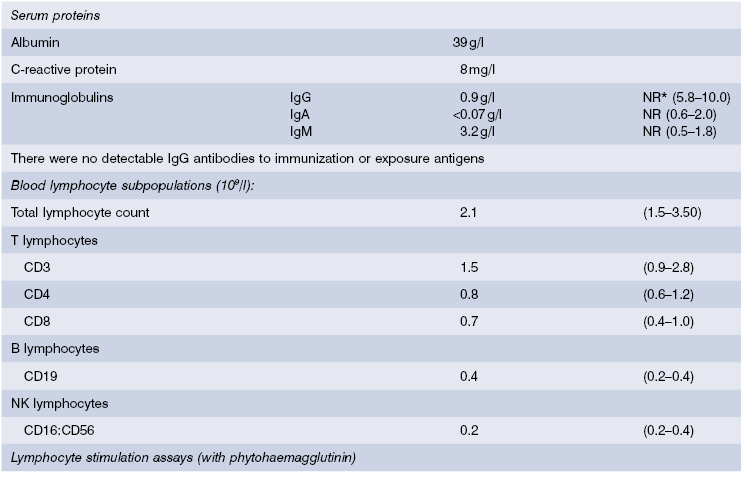
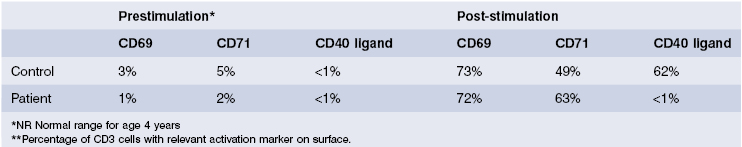
This was most likely to be due to a hyper-IgM syndrome as the serum IgM was raised and cryptosporidia is a particular feature of this condition. Peripheral blood lymphocytes were separated and stimulated in culture; activation markers, including CD40 ligand, were then detected by flow cytometry. CD69 and CD72 were present but there was no CD40 ligand on activated T lymphocytes. Mutation analysis confirmed a deletion in the CD40 ligand gene on the X chromosome and a substantive diagnosis of CD40 ligand deficiency was made. He was treated initially with replacement immunoglobulin, co-trimoxazole to prevent Pneumocystis infection and specific antibiotics for Cryptosporidiosis; if this organism can be controlled, human stem cell transplantation, with or without liver transplantation, will be considered. His mother was tested for carrier status, as will his sisters when they reach the age of consent.
Fig. 3.7 Lymph node from a patient with CD40 ligand deficiency showing impaired germinal centres. (Courtesy of F. Fachetti.)
Management of such patients currently consists of replacement immunoglobulin and genetic testing for potential female carriers. Human stem cell transplantation in childhood is now considered to be the treatment of choice, since a high proportion of patients develop liver disease or malignancies in later life.
Failure of any part of the interaction between CD40 ligand and CD40, or of the other essential components of the pathway (including other ligands and intracellular enzymes involved), results in failure in immunoglobulin switch and germinal centre formation. These pure B-cell forms of the hyper-IgM syndrome include defects in the enzymes responsible for repair of DNA in somatic hypermutation in B cells – such as activation-induced cytidine deaminase (AID). Such patients fail to switch and have a limited antibody repertoire; there is an accumulation of immature B cells in abnormal germinal centres and hence clinically enlarged lymph nodes and spleens.
Common variable immunodeficiency disorders
CVIDs are a heterogeneous group of disorders, making up the commonest form of primary antibody deficiency, an accounting for about 90% of the symptomatic antibody deficiencies. The variable nature of the conditions is reflected in the name. The several diseases with this type of immunological phenotype have become more distinct in recent years. Although some patients present in childhood, most (> 90%) are not diagnosed until adulthood (see Cases 3.3 and 16.7). Most patients with a CVID have low serum levels of IgG and IgA, with normal or reduced IgM and normal or low numbers of B cells. A small group of patients have low circulating T cells as well and have been redefined as ‘late-onset combined immunodeficiency’; many of the patients with complex disease and multiple complications also have abnormal T-cell function, which may reflect an, as yet, undescribed combined immune deficiency (CID). Affected individuals experience the same range of bacterial and viral infections as other patients with antibody deficiencies (see Figs 3.2 and 3.4).
Currently, many patients lead normal lives, provided that they receive replacement immunoglobulin therapy. Complications are very varied (see section 3.2.4), probably due to complication-causing or disease-modifying genes (such as TACI), resulting in the different syndromes in this large group of disorders. Inheritance is very rare (<5%) and affected women have normal offspring, as in Case 3.3.
 Case 3.3 Common variable immunodeficiency disorder (CVID)
Case 3.3 Common variable immunodeficiency disorder (CVID)A 34-year-old woman developed herpes zoster and lobar pneumonia; over the previous 5 years she had been admitted to hospital with pneumonia on two previous occasions and made a full recovery. There had been no history of recurrent chest infections during childhood. Non-encapsulated Haemophilus influenza and Streptococcus pneumoniae were isolated. At the age of 35, she developed a non-erosive seronegative arthritis. On direct questioning, she gave a history of intermittent diarrhoea since her late teens. These episodes lasted from 2 days to 2 weeks and she passed five to six partly formed stools a day. There was no family history of recurrent infections: she had two sons, aged 10 and 7, both of whom were well. Physical examination was normal, although she was thin.
Investigations showed a haemoglobin of 115 g/l, with normal neutrophil and lymphocyte counts. Immunological studies (Table 3.6) showed very low levels of serum immunoglobulins, and no detectable specific antibodies despite culture-proven Streptococcus pneumoniae and a tetanus toxoid boost 1 year earlier. She had normal numbers of circulating T and B lymphocytes. No infective cause of the intermittent diarrhoea was found; barium enema and colonoscopy were normal.
Table 3.6 Immunological investigations* in Case 3.3, a CVID
| Quantitative serum immunoglobulins (g/l): | ||
| IgG | 3.15 | NR (7.2–19.0) |
| IgA | 0.11 | NR (0.8–5.0) |
| IgM | 0.66 | NR (0.5–2.0) |
| Antibody activity | ||
| Post-immunization IgG to: | ||
| Tetanus toxoid | Negative (>0.85 IU/ml) | |
| Diphtheria toxoid | Negative (>0.2 IU/ml) | |
| Pneumococcal polysaccharides | Negative (>80 U/ml) | |
| Blood lymphocyte subpopulations (×109/l): | ||
| Total lymphocyte count | 1.6 | (1.5–3.5) |
| T lymphocytes | ||
| CD3 | 1.31 | (0.9–2.8) |
| CD4 | 0.89 | (0.6–1.2) |
| CD8 | 0.41 | (0.4–1.0) |
| B lymphocytes | ||
| CD19 | 0.2 | (0.2–0.4) |
| NK lymphocytes | ||
| CD16: CD56 | 0.2 | (0.2–0.4) |
*Normal adult ranges shown in parentheses.
She was diagnosed as having a common variable immunodeficiency disorder, a diagnosis of exclusion, as no underlying cause was found. She was given fortnightly intravenous infusions of human normal IgG (400 mg/kg body weight/month) for the antibody deficiency. However three years later she developed pain, bloating and further diarrhoea. Duodenal biopsies showed flat villi without pathogens. A gluten-free diet (to which she adhered rigidly) was not successful in reducing the abdominal symptoms. Ultimately she failed to absorb fat soluble vitamins A, D and E and lost 6 kg in weight. She had the enteropathy associated with CVID, the pathogenesis of which is uncertain. She died suddenly of an unrelated pulmonary embolus.
Selective or partial antibody deficiencies
So called ‘IgG subclass deficiencies’ are controversial, since deletion of IgG subclass genes does not necessarily lead to disease. Investigation of individual patients should be limited to those with significant recurrent bacterial infections. In such patients, the total IgG level may appear normal so selective deficiencies of one or two of the three protective IgG subclasses (IgG1, IgG2 and IgG3) may be missed. However, what really matters is the ability to make specific antibodies against infective organisms to prevent recurrent infections, so reduced IgG subclass levels alone are not always significant. Most clinically relevant deficiencies of IgG subclasses are those associated with IgA deficiency. IgG subclass measurements are only needed if there is also a low serum IgA level, the patient suffers from recurrent infections and also fails to make specific antibodies to some antigens (Case 3.4).
 Case 3.4 IgA with IgG subclass deficiencies
Case 3.4 IgA with IgG subclass deficienciesA 48-year-old man was admitted for investigation of weight loss associated with intermittent diarrhoea; stool examinations had been unhelpful. He had a history of pneumonia as a child and again as a young man working abroad. At the age of 33 he had developed chronic sinusitis, with persistent headaches. On examination, he was thin but had no signs of malignancy. There was no clubbing, lymphadenopathy or hepatosplenomegaly and his chest was clear on auscultation. Haemoglobin, serum albumin, liver function tests and urine electrophoresis were normal. Immunological tests are shown in Table 3.7. Investigations into the cause of the recurrent diarrhoea revealed Giardia lamblia on jejunal biopsy, even though microscopy was negative even though microscopy was negative. Endoscopic examination of his maxillary sinuses showed considerable inflammation and hypertrophy of the mucosa.
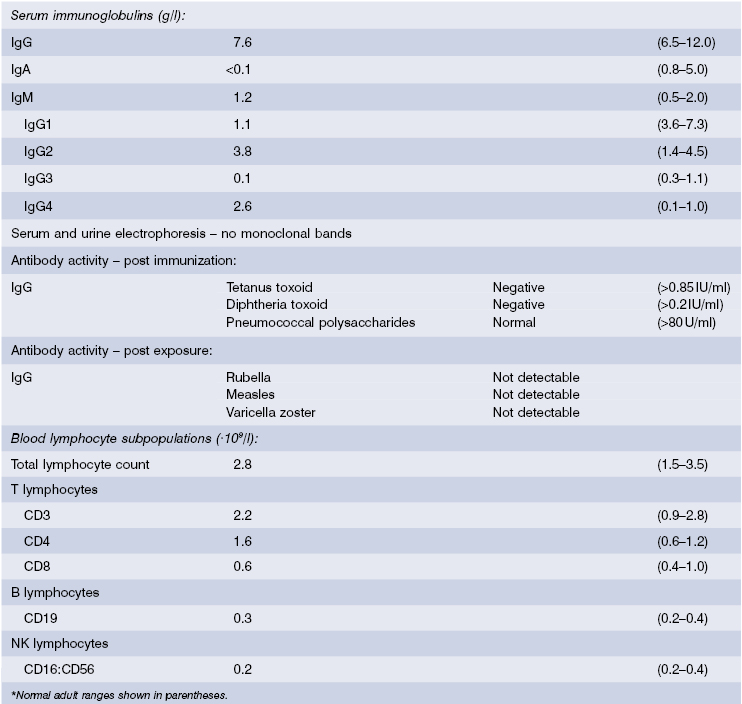
A diagnosis of IgA with IgG subclass deficiencies, with chronic sinusitis and intestinal giardiasis as secondary complications, was made. He was given a course of metronidazole for the giardia infestation and replacement immunoglobulin was started with weekly infusions initially and subsequently 3-weekly at a dose of 0.4 g/kg per month. The sinusitis gradually improved, the diarrhoea did not return and he remained infection free for many years.
Antibodies are produced in different ways to carbohydrate and protein antigens. Antibodies to polysaccharide capsular antigens of organisms, such as those of Streptococcus pneumoniae, Salmonella typhi or encapsulated Haemophilus influenzae (see Fig. 3.2), are often transient, low affinity and of the IgG2 subclass. Those to protein antigens, such as viral coats and toxoids, are usually persistent, high affinity and of the IgG1 subclass.
Polysaccharide antigens alone do not stimulate immune responses in children under 2 years of age, explaining why severe infections with encapsulated organisms were relatively common in infants until the advent of conjugated polysaccharide:protein vaccines. Antibody deficiencies, other than those with a known genetic cause, cannot be diagnosed until children are over 4 years, to allow for physiological variation (see Transient hypogammaglobulinaemia – page 57).
Selective IgA deficiency
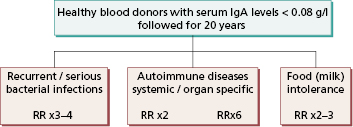
This is the commonest primary defect of specific immunity, with an incidence of 1:700 in Europe, Japan and the USA. It can present at any age, although most patients are diagnosed by an incidental finding as young adults. It is characterized by undetectable or very low serum IgA levels, with normal concentrations of IgG and IgM and production of normal antibodies to pathogens. So most individuals are healthy and do not suffer from recurrent infections.
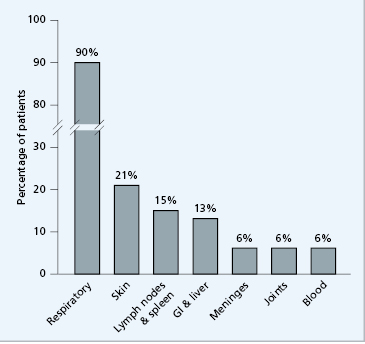
Nevertheless, selective IgA deficiency predisposes the individual to a variety of disorders (Fig. 3.8), in particular coeliac disease (gluten sensitivity).
3.2.3 Differential diagnosis
Primary antibody deficiencies are relatively rare causes of recurrent infections and the differential diagnosis is wide. If infections recur at a single site, then a local cause is likely. For example, cases of recurrent meningitis are usually caused by a passage communicating the ear or sinuses with cerebrospinal fluid, while recurrent pneumonia may be due to structural lung damage or aspiration of a foreign body.
Secondary causes of immunoglobulin deficiency (see section 3.5) are far more common than primary defects. Many textbooks provide long lists of causes. From the practical viewpoint, however, this is not a particularly helpful approach. For example, although the nephrotic syndrome is relatively common in childhood (compared with primary antibody deficiency) and certainly causes isolated low serum IgG levels, recurrent infections are rarely a significant problem, since antibody production is normal.
It should be remembered that patients with primary antibody deficiency can present at any age and a search for an underlying cause for antibody deficiency should always be made in patients with recurrent/severe/persistent/unusual infections (as in Cases 3.1–3.4).
3.2.4 Complications of primary antibody deficiencies
Patients with all forms of primary antibody deficiency suffer from a wide range of bacterial infections (Fig. 3.2). Chronic sepsis of the upper and lower respiratory tracts can lead to chronic otitis media, deafness, sinusitis, bronchiectasis, pulmonary fibrosis and ultimately cor pulmonale. Mild infective gastrointestinal disease occurs in up to two-thirds of patients with primary antibody deficiencies, and in about 20% it warrants further investigations. Diarrhoea, with or without malabsorption, is most frequently caused by infestation with Giardia lamblia, bacterial overgrowth of the small intestine, or persistent infection with cryptosporidium (in those with CD40 ligand deficiency), salmonella, campylobacter, rotavirus or enteroviruses. Chronic cholangitis may be due to ascending infection of the biliary tract; some cases progress to sclerosing cholangitis or hepatic cirrhosis.
Virus infections are rare, but patients with X-linked agammaglobulinaemia or a CVID are susceptible to chronic enterovirus infection. This can result in severe, persistent meningoencephalitis, sometimes with an associated dermatomyositis-like syndrome. Death often follows, despite high doses of immunoglobulin therapy. Ureaplasma may cause genitourinary infections.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


