Earliest definition
Current definition
ECOG’s new definition
Myeloid
Myeloid
Myeloid
HLA-DR NEG
HLA-DR NEG
HLA-DRNEG
CD34 HLA
CD34 NEG
CD11a NEG
CD15 NEG
CD18NEG
CH15s POS
CD133 NEG
CD9 POS
CD45 WEAK
CD38 LOW
CD38 WEAK
Pgp WEAK/NEG
CD15 NEG
The (15;17) translocation involves the retinoic receptor α (RARA) gene on the long arm (q) of chromosome 17 and the promyelocytic leukemia (PML) gene on the q arm of chromosome 15. While the breakpoints in the RARA gene occur consistently in intron 2, differential breakpoints in the PML gene lead to the L- (Long, bcr1), S- (Short, bcr3), or the V- (Variable, bcr2) transcript isoform of PML/RARα. The HLA-DRLOW, CD11aLOW, CD18LOW surrogate marker profile is applicable to all three molecular isoforms [31, 33]. However, the isoforms can be distinguished based on specific antigenic features outside this core antigen profile. While the antigen expression patterns in L- and V-form diseases are indistinguishable, they are clearly separable from S-isoform APL. Only leukemic promyelocytes that contain S-form transcripts variably express CD34, CD2, and CD56. The expression of CD2 together with the S-isoform correlates with poorer prognosis [55]. A potential explanation could be a higher incidence of extramedullary relapse in CD56POS cases [56]. Furthermore, thrombotic events in APL were found to be associated with CD2POS S-form disease with FLT3-ITD [57]. As discussed before, CD2POS APL is hypothesized to be derived from a progenitor cell with myeloid/lymphoid potential [35, 58]. Note, while CD2 and/or CD56 on a patient’s leukemic promyelocytes are highly suggestive of the S-isoform, because they are never seen in L- or V-isoform, there are S-isoform patients who lack these antigens. CD34 expression may be associated with the molecular isoform or the microgranular morphologic variant (see before). Even when expressed, the density of the CD34 antigen is significantly lower on the surface of leukemic promyelocytes than on non-APL myeloblasts [59]. Until now not appreciated is the observation that CD2POS APL occasionally may lack APL-typical antigenic features, something never seen in L- or V-form APL, thus possibly misleading laboratory investigators or pathologists when they interpret the data [3]. It is recommended that any case of CD2POS myeloid leukemia be immediately tested by polymerase chain reaction (PCR) for PML/RARα or CBFβ/MYH11, since APL and inv(16)(p13q22)/t(16;16)(p13;q22) AML (see following discussion in this chapter) are the two major AML subtypes found to express CD2 [4], especially since these leukemia subdiagnoses require distinct therapies.
In addition to the PML/RARα fusion gene, which accounts for >98 % of APL cases, a common segment 5′truncated RARA has been found to fuse with alternative genes [60]. Because such variant translocations are rare, clinical information regarding their responsiveness to ATRA is scarce; recurrent cases of PLZF/RARα (promyelocyte leukemia zinc finger), derived from t(11;17)(q23;q21), appear to lack ATRA responsiveness, while NPM/RARα (nucleophosmin) APL, derived from t(5;17)(q35;q21), is ATRA responsive. Despite their low frequency, the limited immunophenotypic observations available suggest that the main characteristic features of PML/RARα APL cells hold up for all currently known variant APL translocations that involve rearrangement of the RARα gene [31]. Novel variant RARα fusion genes keep appearing in the literature whereby ATRA sensitivity in individual cases appears to vary. Two pieces of evidence should prompt the search for the presence of an alternative RARα fusion gene in a patient: (a) the finding of APL-specific immunophenotypic features in a patient negative for PML/RARα, and (b) cytogenetic evidence of chromosome 17 abnormalities in such a patient. Occasionally, slight variations from the typical APL profile may be found. Gallagher et al. [61], for instance, found weak expression of CD133 in the only second case of STATb/RARα. If confirmed in further cases, this antigenic peculiarity may serve as a diagnostic tool for this particular APL variant.
t(8;21)(q22;q22)-AML1/ETOPOS AML
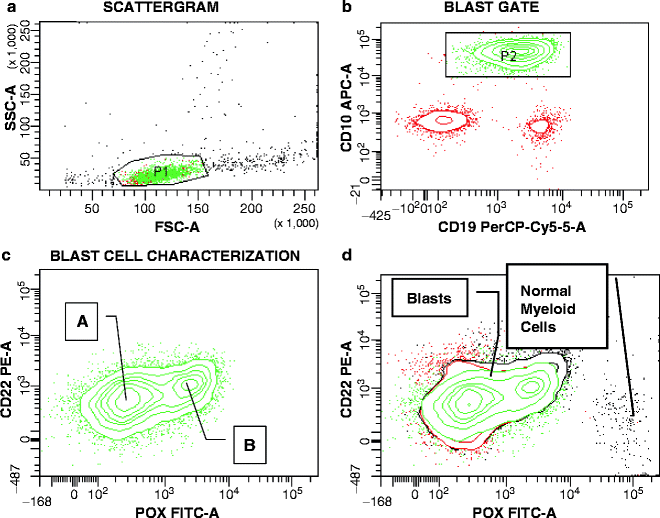
Leukemias with t(8;21) or inv(16)/t(16;16) belong to the core binding factor (CBF) AMLs, which are a diagnostically and prognostically distinct subgroup [9]. Both of these chromosomal rearrangements result in the formation of fusion proteins, AML1/ETO and CBFβ/MYH11, respectively, that involve the disruption of one of the CBF transcription factor genes. The two genes involved in (8;21) translocation are the AML1 transcription factor, now called RUNX1, on chromosome 21q22.3, and the Eight-Twenty One oncoprotein (ETO) (also called MTG8) on chromosome 8q22 [29]. AML1/Runx1, one of three α-subunits of the CBF family, is a sequence-specific DNA-binding protein, while the single CBFβ subunit is non-DNA binding but enhances the DNA affinity of the α-subunits [62]. The characteristic immunophenotype of t(8;21) AML allows for a correct prediction of this genetic aberration [63]. The initially recognized distinctive features were expression of CD19, a B-lineage associated antigen, and of CD56, the neural-cell adhesion molecule, by CD34POS myeloblasts. Presence of CD56 may explain the increased incidence of granulocytic sarcomas observed in this disease [64, 65]. While CD19 is consistently present in the t(8;21) subtype and rarely found in other AMLs, expression of this antigen by myeloblasts is often very weak so that its detection can depend on the accurate choice of fluorochromes and an open mind on the part of the interpreting flow cytometrist. In fact, CD19 conjugated to fluorescein isothiocyanate (FITC) should be avoided at all costs. An example of CD19/CD56 double expression in a patient with AML1/ETOPOS AML is shown in Fig. 17.1. Although CD56 expression is promiscuous among myeloid leukemias and absent in a marked fraction of t(8;21) cases, the finding of both CD19 and CD56 against the background of a myeloid phenotype is highly suggestive of t(8;21) AML. As in the case of APL, an increasingly accurate surrogate marker profile for t(8;21) AML has evolved over time. One particularly helpful diagnostic tool is the diminished or absent expression of CD11a/CD18 [31, 66], a member of the β2 integrin subfamily [42]. With the exception of t(8;21) or t(15;17) (and its variants), >90 % of AML demonstrate expression of CD11a/CD18, albeit often with low intensity. The absence of CD11a in t(8;21) AML is explained by the inhibition of Runx1-dependent CD11a transcription by the AML1/ETO fusion product [66]. Variable expression of CD11a is seen in AML with dual expression of CD19 and CD7, a T-lineage affiliated antigen [48]. Previously, the incompatibility of t(8;21) with expression of CD7 had been reported [67]. More recently, CD19/CD7 double positive AML has been associated with a predominantly normal karyotype and FLT3-ITD and NPM1 mutations [50, 68].
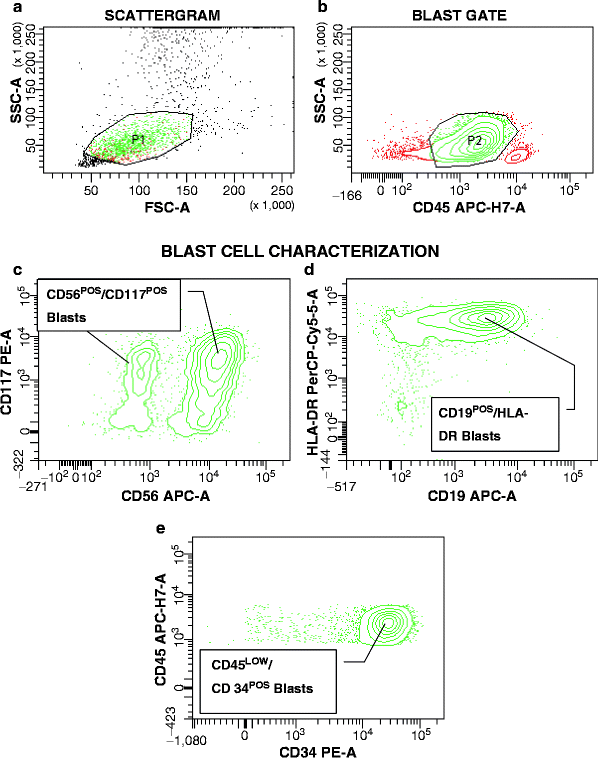
Fig. 17.1
Dual CD19 and CD56 expression by t(8;21)-AML1/ETOPOS myeloblasts. (a) P1 is drawn around suspicious cells with low side (SSC) and forward scatter (FSC), reflecting lack of granularity and small to intermediate cell size. (b) In this biparametric contour plot, cells within gate P1 are found to contain a major population with low CD45 expression (gate P2, green contours), clearly separated from CD45HIGH normal lymphocytes. (c–e) CD45LOW cells are further characterized and found to stain with CD117, CD56, HLA-DR, CD19, and CD34, consistent with leukemic myeloblasts with immunophenotypic characteristics of t(8;21)-AML1/ETOPOS disease. Based on CD117 versus CD56 (c), two blast cell populations are distinguished differing in the fluorescence intensity of CD56 antibody staining. The intensity of CD19 and CD117 staining is relatively homogenous with a smaller number of blasts displaying reduced density of expression of both antigens. HLA-DR and CD34 are both strongly expressed on the leukemic myeloblasts. The blasts also demonstrated low expression of CD11a/CD18 and CD33 (not shown), further confirming the surrogate marker profile for t(8;21)-AML1/ETOPOS AML
Aside from CD19, t(8;21) myeloblasts can also express two other B-lineage antigens, CD79a and PAX5 [30]. The PAX5 gene encodes a paired box domain transcription factor, which is considered a crucial mediator of B-cell identity [69]. Tiacci et al. [30] hypothesized that the PAX5-dependent expression of CD19 and CD79a in t(8;21) AML results from the interaction and functional cooperation between the PAX5 and AML1/ETO proteins. Expression of CD56 is also not limited to the myeloid lineage [70] and in B-lineage ALL appears to be associated with BCR/ABL transcripts [71]. Taken together, these findings can lead to a misdiagnosis of B-lineage ALL, especially in view of the immaturity of the myeloid phenotype frequently encountered (as discussed before), as long as karyotypic and molecular data are unavailable. While in t(8;21) AML, the same blast population will co-express myeloperoxidase or surface myeloid antigens, e.g., CD13, weak CD33, and CD19, CD79a, PAX5, these blasts will not stain for cCD22. It is important to remember that dual absence of myeloperoxidase and cCD22 is consistent with AML. Given that the evaluation of intracytoplasmic antigens continues to be a challenge for many flow cytometric laboratories, this is a typical situation, in which the rapid flow cytometric assessment of AML1/ETO protein, anticipated for the near future [72], will be of invaluable help in the correct diagnosis of t(8;21) AML.
inv(16)(p13q22)/t(16;16)(p13;q22)-CBFβ/MYH11POS AML
The expression of the T-cell affiliated antigen CD2 by leukemic myeloblasts is rarely observed, provided that antigen expression is viewed on gated leukemic cells exclusively. Although not (yet) part of a full surrogate marker profile, single antigens can serve as “diagnostic pointers” for genotypes. CD2 expression by leukemic cells with a myeloid phenotype should raise the immediate suspicion of APL (see previously in this chapter). The other genotypically defined AML subtype that frequently demonstrates CD2 expression is CBFβ/MYH11POS AML derived from inv(16)(p13q22) or t(16;16)(p13;q22) [73]. APL and CBFβ/MYH11pos AML can be easily distinguished given the characteristic APL immunophenotype (HLA-DRNEGCD11aNEGCD18NEG) that is distinctly different from that of CBFβ/MYH11POS disease.
Analogous to CD19 in t(8;21) leukemia, CD2 expression by CBFβ/MYH11POS can be very weak and may be overlooked, definitely if FITC-conjugated CD2 is tested. Occasionally, however, CD2 expression can be quite strong, as exemplified in Fig. 17.2. This is an important, though not well-appreciated association, given that inv(16) is a subtle cytogenetic abnormality that is easily missed. The expectation of the typical morphologic picture associated with CBFβ/MYH11POS AML, FAB M4 and abnormal eosinophils (FAB 4eo), leads pathologists to shy away from investing efforts into a refined surrogate marker for this AML subtype. However, central morphologic review of 98 CBFβ/MYH11POS ECOG AML patients demonstrated that FAB M4eo was confirmed in only 59 % of cases, followed by M4 without eosinophilia (13 %), M2, with or without eosinophilia (19 %), M1 (5 %), and M5 (3 %) (Bennett JM, personal communication). When compared with institutional pathology reviews, the concordance with FAB M4eo was even lower, suggesting that immunophenotypic assistance in the diagnosis of this AML subtype could be useful.
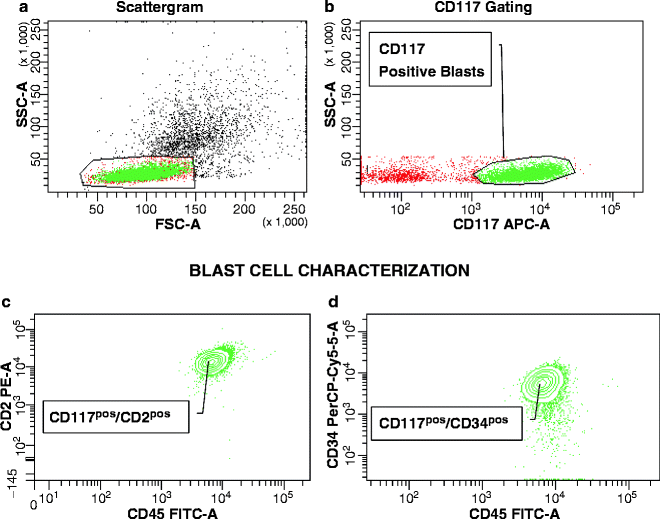
Fig. 17.2
CD2 Expression in a patient with inv(16)-CBFβ/MYH11POS AML. (a) Based on light scatter characteristics (granularity and size), a gate is set around suspicious cells (P1). (b) In this plot, the majority of events gated in P1 are found to express CD117 (P2, green dots), identifying these cells as abnormal. Further characterization of cells in P2 in contour plots (c) and (d) reveal strong expression of CD2 and CD34, typical features of inv(16)-CBFβ/MYH11POS AML
t(9;22)(q34;q11)-BCR/ABLpos ALL
The Philadelphia chromosome, t(9;22), which results in the BCR/ABL fusion gene, is the dominant negative prognostic factor in ALL [74]. Its incidence increases with age, accounting for 2–5 % in children and up to 28 % in adult patient cohorts [75]. The disappointing response of BCR/ABLPOS patients to standard therapy and the availability of specific inhibitors for the constitutively activated tyrosine kinase in BCR/ABL fusion proteins [76] have prompted separate trials for BCR/ABLPOS and BCR/ABLNEG ALL, making it imperative that these patients are recognized accurately.
Cytogenetic analyses in lymphoblasts are hampered by suboptimal chromosome structure [77], and, furthermore, approximately 10 % of BCR/ABLPOS ALL lack evidence of the t(9;22) by chromosome banding [75]. Both karyotyping and molecular analyses by PCR require time, 1–2 weeks and 48 h, respectively. The flow cytometric bead assay for BCR/ABL fusion proteins [72], that is discussed next, is the ideal solution, though an expensive one. Long before the development of this bead assay, there was intense interest and urgency in the establishment of a surrogate marker profile for BCR/ABLPOS ALL. The first alleged antigenic signature relied solely on differences in staining intensity of antigens commonly found in B-lineage ALL, such as strong expression of CD10 and CD34 and weak expression of CD38 [78–80]. However, in 1997, a preliminary analysis of 144 patients enrolled in ECOG’s phase III adult ALL trial, E2993, demonstrated an association between BCR/ABL positivity and expression of CD25, the α-chain of the interleukin-2 receptor [81]. This observation confirmed an earlier report of the incidental finding of CD25 in four patients with BCR/ABLPOS ALL [82]. The final analysis of E2993 [83] solidified the surrogate marker profile for BCR/ABLPOS lymphoblasts as CD25posCD34high with dual expression of myeloid antigens, CD33 and CD13. Despite the frequent expression of two myeloid antigens by BCR/ABLPOS lymphoblasts, this phenotype must not be considered biphenotypic, since BCR/ABLPOS lymphoblasts unequivocally belong to the B-cell lineage (cCD22POS, myeloperoxidaseNEG). Greater than 75 % of BCR/ABLPOS lymphoblasts express additional cytogenetic aberrations [83]. Primo et al. [84] reported that patients with del(9)(p21), in addition to the t(9;22), lacked both CD33 and CD13. Among 11/156 BCR/ABLpos patients with del(9)(p11), del(9)(del(9)(p13), or del(9)(p21) on E2993, 8 lacked both CD33 and CD13, whereas the other 3 only showed decrease or loss of CD13 expression, thus supporting and expanding Primo’s observation (Paietta E, personal observation).
The dual expression of myeloid antigens, CD33 and CD13 by BCR/ABLPOS lymphoblasts, is not surprising, given that these blasts most commonly express immunophenotypic features of early Pre-B ALL (CD10POS). Ludwig et al. [85] have proposed that the expression of myeloid antigens in adult B-lineage ALL differs according to the level of B-lymphoblast maturation, analogous to what has been reported from pediatric ALL [86, 87]. Preliminary analysis of the largest adult ALL trial to date (ECOG 2993) [88] confirmed but also expanded these associations. While Pro/Pre-Pre-B ALL, the immature CD10NEG maturation stage, typically expressed CD65(S) and CD15(S), the CD10POS early Pre-B stage preferentially expressed the pan-myeloid antigens CD33 and CD13, which in normal myelopoiesis appear before CD65(S)/CD15(S) on maturing myeloid cells. However, these relationships did not hold up in BCR/ABLPOS cases; as given in Table 17.2, the paired expression of CD33/CD13 persisted irrespective of the maturation stage of BCR/ABLPOS lymphoblasts. CD33/CD13 positivity was seen whether BCR/ABLPOS blasts lacked CD10 (Pro/Pre-Pre-B stage) or expressed intracytoplasmic μ chains (Pre-B stage). Importantly, BCR/ABLPOS lymphoblasts never expressed CD65(S)/CD15(S) even in those instances in which CD33 and/or CD13 were not expressed.
Table 17.2
Numbers in bold and italic represent the high frequency of CD33/CD13-dual positive B-lymphoblasts in BCR/ABLPOS B-lineage ALL, irrespective of the maturation stage of the disease. Italic numbers (not bold) indicate differential myeloid antigen expression in BCR/ABL negative disease by B-ALL maturation
BCR/ABL | B-ALL maturation | CD33 | CD13 | CD65 | CD15 |
|---|---|---|---|---|---|
Negative | Pro-B /Pre-Pre-B | 20 | 20 | 57 | 72 |
Positive | Pro-B/ Pre-Pre-B | 40 | 50 | 0 | 15 |
Negative | Early Pre-B | 45 | 49 | 7 | 14 |
Positive | Early Pre-B | 72 | 81 | 1 | 10 |
Negative | Pre-B | 3 | 16 | 13 | 29 |
Positive | P | 64 | 82 | 0 | 9 |
The (9;22)(q34;q11) translocation results in the actual Philadelphia chromosome, the derivative chromosome 22, in which the BCR/ABL fusion gene is located, and the derivative chromosome 9, where the ABL/BCR fusion gene resides, which does not appear to contribute to the pathogenesis of this disease. Various isoforms of the BCR/ABL fusion gene are created dependent on the variable breakpoints in the BCR gene. All translated BCR/ABL proteins share a similar carboxy terminal ABL tyrosine kinase domain (TKD), but differ in the portion of the BCR protein included in the fusion product, due to multiple breakpoint cluster regions in the BCR gene. In the majority of chronic myeloid leukemia (CML) and in one third of BCR/ABLPOS ALL, the break within the BCR gene occurs in the major breakpoint cluster region (M-bcr), resulting, when joined with a portion of c-ABL from chromosome 9, in a b2a2 or b3a2 fusion transcript encoding a protein of 210,000 Da molecular weight (p210BCR-ABL). A break in the minor breakpoint cluster region (m-bcr) forms the e1a2 transcript encoding a 190,000 Da protein (p190BCR-ABL), found mostly in BCR/ABLPOS ALL [89, 90]. The antigen expression profile for BCR/ABLPOS ALL varies with the BCR/ABL isoform, in that dual myeloid antigen expression of CD33+CD13 and CD25 expression are much more frequent in p210BCR-ABL lymphoblasts [83].
The most striking observation with respect to CD25 and BCR/ABLPOS ALL is that expression levels of CD25 are of prognostic significance predicting for a lower likelihood to achieve complete remission [81, 83], and shorter overall (OS) [81, 83] or event-free survival [91] among BCR/ABLPOS patients. In other words, CD25 is unique in its dual function as a dependable marker of BCR/ABLPOS ALL and an independent prognostic factor for outcome in this disease.
Though isolated case reports of BCR/ABLPOS ALL with T-lineage phenotype are still occasionally posted, all of the larger studies have reported that BCR/ABL rearrangements in pediatric and adult ALL are restricted to the B-cell lineage [76, 92–94]. A substantial cohort of pediatric patients studied from various European groups found 3 T-ALLs among 61 BCR/ABLPOS patients [95]; unfortunately, no details were provided on the immunophenotypic diagnosis of those T-lineage cases, though it is apparent that cCD3 and cCD22 were not tested. The lack of demonstration of these lineage-specific antigens is the most likely reason for misdiagnosed BCR/ABLPOS T-cell phenotypes. A potential alternative cause is an apparent Philadelphia chromosome translocation by conventional karyotyping that does not yield the ABL-BCR juxtaposition on chromosome 9 [96]. While in the report by Fossa et al. [96] the leukemic phenotype was unequivocally T-lymphoid (cCD3POS), an analogous case was seen in ECOG’s E2993 trial, with a (9;22)(q34;q11.2) translocation that by fluorescence in situ hybridization (FISH) did not result in the BCR/ABL fusion but, as in Fossa’s case, disrupted the BCR gene (ECOG Cytogenetics Committee, personal communication). The E2993 patient, however, expressed immunophenotypic features of Transitional Pre-B ALL. In summary, caution is definitely warranted when t(9;22)POS or BCR/ABLPOS T-ALL cases are published.
t(4;11)(q22;q23)-MLL/AF4pos B-Lineage ALL
MLL/AF4 is the typical subtype of infant ALL and the most prevalent of the diverse mixed lineage leukemia (MLL) fusion genes in adult ALL [97–99]. Despite improved complete remission rates with modern treatments, overall survival in adults remains poor due to short remission duration [98, 99]. MLL/AF4POS ALL is associated with the immature Pro/Pre-Pre-B ALL maturation stage, defined by negativity for CD10. As a unique immunophenotypic feature among all ALL phenotypes, MLL/AF4POS lymphoblasts show a tendency to express the more mature myeloid carbohydrate antigens, CD65(S) and CD15(S), while lacking CD33 and CD13, the pan-myeloid antigens expressed earlier in normal myelopoiesis [71, 99, 100]. On the other hand, CD33 and/or CD13 are found preferentially in CD10pos early Pre-B ALL. Burmeister et al. [100] reported that lymphoblasts from MLL-rearranged adult ALL patients (55 % with MLL/AF4) demonstrated significantly lower expression of CD10, CD33, and CD13 and more frequent expression of CD65(S) and CD15(S) irrespective of B-lineage differentiation stage. Together with the myeloid antigen expression pattern in BCR/ABLPOSALL [88], these observations suggest that myeloid antigen expression in B-lineage ALL is determined by the underlying genetic defect rather than B-lymphoid developmental stage.
Cryptic t(12;21)(p13;q22)-TEL/AML1 B-Lineage ALL
The cryptic (12;21)(p13;q22) translocation that results in the TEL/AML1 fusion gene is the most common genetic aberration in pediatric ALL (>20 %) and carries a favorable prognosis, especially with high-intensity therapy [101, 102]. In adults, this hybrid gene only accounts for <1–3 % of ALL [80, 103, 104], thus precluding prognostic predictions. In children, the TEL/AML1 fusion occurs exclusively in Early Pre-B ALL with certain characteristic markers: high-intensity of HLA-DR, CD40, and CD10 expression (Early Pre-B phenotype), commonly CD13POSand/or CD33POS, but CD9NEG, CD20NEG, and low expression of CD45, CD135, and CD86 [105–107]. A striking characteristic feature has recently been added, namely absence of CD11b expression [108]. Given the low incidence of TEL/AML1POS ALL in adults, immunophenotypic information in this age group is scarce. The nine TEL/AML1POS cases found among ECOG E2993 patients (1.4 %) demonstrated the characteristic CD10POS Early Pre-B phenotype, lacking CD20 and predominantly CD34, and frequently expressing CD33+CD13. However, in comparison to Early Pre-B ALL with normal cytogenetics and molecularly negative for TEL/AML1, BCR/ABL, MLL/AF4, and E2A/PBX1, there was no difference in CD45, and the CD40 intensity was higher than the median value in normal karyotype controls in only half of the patients [104]. Furthermore, CD11b was expressed by all lymphoblasts in two of the seven TEL/AML1POS E2993 cases, in which this antigen was tested (Paietta E, unpublished observation). Although based on small numbers of patients, the most predictive immune profile applicable for both pediatric and adult TEL/AML1POS B-lineage ALL, to date, is CD10POS,CD20NEG, CD34NEG, cIgMNEG, frequently CD33+CD13POS, and possibly CD11bNEG.
Inv(7)(p15q34) and t(7;7)(p15;q34)-TRB@/HOXA
As a result of two cryptic cytogenetic aberrations, inv(7)(p15q34) or t(7;7)(p15;q34), the TCRB locus (TRB@) at 7q34 is juxtaposed to the HOXA@ at 7p15 (approximately <5 % of pediatric and adult T-ALL), leading to transcriptional activation of several HOXA genes [109, 110]. The typical antigen expression patterns associated with the TRB@/HOXA rearrangement include negativity for CD2 and single expression of CD4, without CD8.
Predictive Antigens for Prognostic Gene Expression
FLT3 Mutations in AML
There are two major clusters of mutations of the FMS-like tyrosine kinase-3 (FLT3) gene, those in the juxtamembrane domain that lead to internal tandem duplications (ITD) and point mutations in the TKD. Both result in the activation of the transforming potential of FLT3 [111, 112]. Activating mutations of the FLT3 gene are the most common known genetic abnormalities in pediatric and adult AML, with FLT3-ITD found in approximately 20–35 % of adults and 15 % in children (ranging from 1.5 % in infants to 20 % at teenage age), while FLT3-TKD are present in about 7 % of AML irrespective of age group. With respect to cytogenetic links, distinct differences are seen between FLT3-ITDs and FLT3-TKDs; unlike FLT3-ITDs, which are particularly frequent in AML with normal cytogenetics and t(6;9)-DEK/CAN, FLT3-TKDs are more common in cases with inv(16)-CBFβ/MYH11. Both types of mutations are found in a large percentage of t(15;17)-PML/RARα APL and are underrepresented in complex karyotypes, 11q23 aberrations, and other adverse cytogenetics [111, 113, 114]. In APL, gene expression profiling clearly distinguished two subtypes based on FLT3 mutation status [35, 115]. In non-APL AML, suggested negative clinical implications for FLT3-ITD (e.g., increased relapse rate) vary with therapeutic intensities, allelic ratio, and size of ITD, and FLT3 with length mutations has become a preferred therapeutic target in AML [116]. FLT3-TKDs, on the other hand, failed to confer unfavorable prognosis; in fact, patients with high-level mutations (more than 25 % mutant) experienced improved outcome [113].
An attempt to establish a unique immunophenotype for FLT3-ITDPOS AML was rather unsuccessful. Kussick et al. [117] suggested a combination of increased CD123, decreased CD38, “mildly” decreased CD117, and loss of CD133 as characteristic of FLT3-ITDPOS AML. Subsequently, the same authors described the distinctive, cup-like nuclear morphology in FLT3-ITDPOS AML (which was discussed before) in association with loss of HLA-DR, CD34, and CD133 [46]. A recent, as yet unpublished, analysis of immunophenotypic features of FLT3-ITDpos AML did not confirm the loss of HLA-DR [46] or CD38 [117] and revealed that lack of CD34 and CD133 expression in FLT3-ITDPOS AML was restricted to patients with concomitant NPM1 mutations (Paietta E, personal observation). Falini et al. reported this paired absence of CD34/CD133 in NPM1-mutated AML in their groundbreaking publication in 2005 [118]. This failed description of a surrogate marker profile for FLT3-ITDPOSAML teaches two important lessons: (1) The major caveat associated with the description of surrogate marker profiles is a reliance on commonly occurring antigens. This is true especially when slight changes in antigen density are considered, which are reflected by weaker intensity of antibody staining and dependent on the choice of fluorochrome. (2) It is only reasonable to assume that the primary genetic lesion in a leukemic cell will determine the phenotype. This assumption is supported by the fact that in NPM1-mutated AML, leukemic myeloblasts lack CD34 and CD133 whether the cells contain only the NPM1 mutation or both NPM1 mutation and FLT3-ITD (Paietta E, personal observation). Because FLT3 gene mutations are secondary rather than primary genetic aberrations, establishing a marker profile specific for this mutation may not be feasible.
FLT3-ITDpos T-Lineage ALL
The rare subtype of CD117POS T-lineage ALL with activating FLT3 gene mutations [11, 119] belongs to the few known examples of gene mutations, which are predicted by the presence of a unique antigen expression pattern.
In pediatric ALL, FLT3-TKDs are common in cases with MLL rearrangements and those with hyperdiploid karyotype, while FLT3 length mutations are extremely rare; still, high levels of FLT3 protein expression provide a target for therapeutic intervention [111, 120]. FLT3 mutations are even rarer in adult ALL. The only large study published to date revealed a 0.7 % incidence of FLT3-ITD among 449 patients [11]. Following completion of the study, the overall incidence of FLT3-ITDs among 511 patients was 1.9 % [119]. Of the ten FLT3-ITDPOS patients, three belonged to the B- and the remaining seven to the T-cell lineage. No consistent immunophenotypic or cytogenetic features were shared by the three Early Pre-B patients. On the other hand, all FLT3-ITDPOS T-ALLs expressed a unique immunophenotype: CD5NEG, surface CD3NEGCD4NEGCD8NEG (triple-negative), positive for CD117, CD34, CD2, CD7, TdT, CD62L, CD13, CD135 (FLT3 protein), and positive for T-cell lineage-specific cCD3 [11]. The complete lack of CD5 expression must be particularly emphasized since it is unique to this subtype of T-lineage ALL. CD117, the stem cell factor receptor, is much more frequently expressed by leukemic myeloblasts than lymphoblasts [121, 122]. In normal lymphopoiesis, a fraction of CD3/CD4/CD8-triple negative, CD34POS thymocytes express high levels of CD117 [123, 124]. In these thymocytes, expression of CD117 coincides with that of CD135, the FLT3 receptor tyrosine kinase [110, 125]. Occasionally, up to 10 % of cCD3POSCD117POSFLT3-ITDPOS blasts expressed myeloperoxidase, thus representing truly biphenotypic features [11]. This profile fits the most immature category of T-ALL [10, 126, 127], resembling earliest thymic precursors with both T- and myeloid lineage potential [128]. In further support of a T-lineage affiliation, FLT3-mutated T-lymphoblasts overexpressed LYL1 and LMO2 oncogenes [11]. In pediatric T-ALL, the LYL1-overexpressing, most immature cases demonstrate relative resistance to standard chemotherapy [10]. To the contrary, the FLT3-ITDPOSCD117POSCD13POS T-cell phenotype in adults does not carry inferior prognosis [119]. This observation is remarkable given the negative prognostic impact of CD13 expression in CD117NEG adult T-lineage ALL [119]. Despite the low frequency of FLT3 gene mutations in adult ALL overall, the availability of a variety of FLT3-kinase inhibitors suggests a potential targeted approach in the treatment of this patient cohort.
Van Vlierberghe et al. [129] subsequently reported that in pediatric T-ALL CD117 mRNA (not protein) expression was not invariably associated with FLT3 gene mutations in that most pediatric cases expressed CD117 mRNA; this suggests that, similar to what has been reported for myeloperoxidase transcripts [130], CD117 mRNA might undergo posttranscriptional downregulation in ALL. Immunophenotypic profiles of the two pediatric FLT3-ITDPOS T-ALL patients [129] were surface CD3NEG but CD5POSCD4POS with partial CD13 expression. Furthermore, the authors stated that these blasts carried a “HOX11L2 translocation,” presumably leading to aberrant HOX11L2 expression. Taken together, these findings suggest arrest at an immature, though already single CD4pos differentiation level, distinct from the triple negative stage seen in the adult cases [11]. Remarkably, another pediatric T-ALL case with FLT3-ITD, the only one among 59 children tested, identified by Ferrando et al. [10] belonged to the LYLPOS cluster, consistent with the findings in the adult cases.
CD117pos T-ALL lacking FLT3 gene mutations were found in 2 % of patients on E2993 [71]. Aside from CD117, their immunophenotype differed from that in FLT3-ITDPOS cases subtly but distinctly: the T-lymphoblasts expressed CD5, occasionally were single CD8POS, frequently expressed CD33 instead of CD13, often were CD56POS, and commonly lacked CD34 and CD62L, both markers of immaturity. A close association between CD117 and CD13 expression in surface CD3NEG T-ALL has been previously reported [131]. Thus, while CD117 expression in adult T-lineage ALL might be considered a surrogate marker for FLT3 gene mutations, this concept only applies for T-lymphoblasts arrested at the most immature stage of differentiation.
NPM1-Mutated AML
The morphologic and immunophenotypic characteristics of NPM1-mutated AML have been mentioned before. In summary, this AML subtype is characterized by high frequency of FLT3-ITD, association with normal karyotype, and negativity for both CD34 and CD133 [48]. However, an informal analysis of the incidence of CD34POS myeloblasts in 105 cases of NPM1-mutated AMLs suggested greater variability than previously appreciated with the percentage of CD34POS blasts ranging from 0 to 99 % (Paietta et al., unpublished observation). Along the same line, LICs in NPM1-mutated AML have been located both in CD34POS and CD34NEG stem cell fractions [132]. Furthermore, the rare immunophenotypic subset of NPM1-mutated/FLT3-ITDPOS normal karyotype-AML with dual CD7/CD19 expression [50] presents with surprisingly high CD34 expression, higher than found in NPM1-mutated/FLT3-ITDPOS AML without this antigen profile or NPM1-mutated AML without FLT3-ITD [68]. To date, no other gene mutations have been identified in the CD7/CD19pos cohort that could explain this change in immunophenotype [68]. Taken together, these observations suggest the existence of heterogenous LICs with distinct properties within and among NPM1-mutated AML cases.
BAALC and/or ERG Expression in T-Lineage ALL
The brain and acute leukemia, cytoplasmic (BAALC) gene is highly expressed in normal uncommitted hematopoietic progenitors and progressively downregulated with differentiation [133]. Low expression of BAALC and of ERG (v-ets erythroblastosis virus E26 oncogene homolog), an oncogenic ETS transcription factor expressed during early T-cell development [134], identified an adult T-ALL subset with highly favorable outcome, particularly lower relapse rate and longer overall survival, and characterized by a predominantly thymic, CD1aPOSCD4POSCD8POS immunophenotype. On the other hand, high expression of BAALC/ERG was associated with high relapse rate, shortened overall survival, and an immature T-cell immunophenotype, negative for CD1a, surface CD3, and occasionally CD5. Furthermore, blasts with high BAALC and/or ERG expression were characterized by CD34 positivity and expression of myeloid antigens, CD33 and/or CD13. In the patient cohort studied, only the immunophenotype and expression levels of BAALC/ERG were of independent prognostic relevance [135]. Of clinical importance, the BAALCLOWERGLOW group comprised 41 % of all T-ALL patients, hence a large subgroup in which excellent long-term outcome could be achieved with chemotherapy alone.
These data are of great interest in view of the recently published immunophenotypic classification of adult T-ALL from E2993, which divided patients into two prognostic subgroups, CD1aPOSCD13NEG and CD1aNEGCD13POS [119]. The CD1aPOS cortical or thymic subtytpe (or Pre-αβ stage according to the TCR-based classification) [127] accounted for 38 % of patients, which lacked CD34 and experienced significantly longer survival than the CD13POS subtype, which typically expressed immature features. While co-expression of CD33 was seen in half of the CD13POS patients, the presence of CD33 was without additive inferior prognostic effect on outcome. Undoubtedly, these CD13POS(CD33POS) T-lymphoblasts [119] strongly resemble T-ALL cases with BAALCHIGHERGHIGH expression [135]. In further support of a common prognostic subgroup, CD1aNEG patients in E2993 had increased risk of relapse [119], an experience shared by the BAALCHIGHERGHIGH group [135]. The Pre-αβ stage [127] was similarly found to be very sensitive to induction therapy and experience superior overall survival [136].
Coustan-Smith et al. [12] described the poor prognostic early T-cell precursor (ETP) ALL in a subset of children with high risk of remission failure or relapse. Notably, ETP-ALL occurred predominantly in children ≥10 years old, demonstrated high ERG, LMO2 and LYL1 expression and had the characteristic immunophenotype: CD1aNEGCD5WEAKCD8NEG, frequently expressing stem cell markers CD34, CD117, and HLA-DR, and one or more of the myeloid antigens, CD13, CD33, CD65, or CD11b. Weak CD5 expression was strictly defined as <75 % CD5POS lymphoblasts. ETP-ALL cases had highly variable cytogenetic abnormalities, including del(5q) and del(13q). This entity probably overlaps with the LYL1 overexpressing most immature T-ALL stage previously associated with poor prognosis [10]. In the TCR-based classification, IM T-ALL represents this immature profile that resembles multipotent thymic progenitors [136].
While it is tempting to relate these poor-prognostic immature T-cell phenotypes to each other, the ETP-ALL [12] in children and the BAALCHIGHERGHIGH subtype [135] and/or CD1aNEGCD13POS T-ALL [119] in adults can only be compared if certain pieces of missing information are provided, such as BAALC and LYL1 expression levels. What can be taken away from this discussion today is that negativity for CD1a, positivity for CD34, and presence of myeloid antigens combined confers inferior prognosis in T-ALL of all ages.
Recently, Gutierrez et al. [137] tested whether their newly discovered poor response pediatric T-ALL subtype with absent biallelic TCRγ deletion had overlapping immunophenotypic characteristics with ETP-ALL [137]. Surprisingly, none but one of the patients demonstrated the low expression of CD5 required for the ETP signature [12]. This discrepancy was eventually resolved and attributed to differences in the interpretation of the low, albeit persistent expression of CD5 by ETP blasts in distinct laboratories [137]. This is an instructive example of the importance of describing surrogate marker profiles with the utmost accuracy in terms of percentage of positive blasts or intensity of staining, fluorochromes chosen, and interpretations applied, if these antigen profiles are to be reproduced by others, especially in multi-institutional analyses.
TLX1/HOX11 Overexpression in T-Lineage ALL
Remarkably, Baldus et al. could not demonstrate differential expression of T-lymphoid transcription factors TLX1/HOX11 in BAALCHIGHERGHIGH versus BAALCLOWERGLOW expressing cases [135], although HOX11 expression has been previously associated with CD1a and with favorable outcome in pediatric and adult T-ALL [10, 13, 138–140], and overexpression of this transcription factor can be detected even in the absence of cytogenetic abnormalities involving the chromosomal regions that contain its gene [141]. The relative frequency of CD1aPOS TLX1/HOX11-induced T-ALL cases appears to be higher in adults than in children [142], which may explain the relatively better outcome for CD1aPOS T-ALL in adults [119] than children [143].
Antibody-Based Detection of Leukemia-Specific Proteins
Up until recently, the detection of disease-specific fusion proteins or gene mutation products in hematologic malignancies was left to molecular testing mostly due to impracticality for antibody-based techniques in routine laboratories or the unavailability of proper antibodies. Nowadays, immunohistochemistry has replaced molecular studies for some proteins, e.g., the ALK protein derived from the (2;5) translocation in various lymphomas [6]. For some proteins, transformation into a hybrid state (e.g., PML to PML/RARα) or mutation (e.g., NPM1 to mutated NPM1) results in the altered distribution within the cell, which can be exploited by recognizing specific immunohistochemical staining patterns [6]. Recently, monoclonal antibodies for flow cytometry have been introduced that either solely recognize mutated NPM1 [144] or the aberrant ectopic cytoplasmic accumulation of the mutant protein [145].
The most exciting development in this area came last year when the Euroflow Consortium in cooperation with the Beckton-Dickinson company developed a commercial kit for the flow cytometric detection of all variants of BCR/ABL proteins encountered in ALL and CML [7]. Although simple in theory, in practice, the detection of fusion proteins turned out to be much more complicated than anticipated [72]. A new system had to be developed, which did not target the fusion protein itself but labeled each part of the protein (e.g., BCR protein and ABL protein) separately. Brilliantly, the investigators coupled a catching antibody to the BCR protein and a phycoerythrin (PE)-conjugated detecting antibody to ABL. To ensure the recognition of all BCR/ABL fusion protein variants, the antibodies were designed against unique epitopes away from all known breakpoints in each gene. The simplicity and reliable performance of this test system and the intentions of this group to expand their methodologic approach to the clinically most significant fusion proteins (e.g., PML/RARα, AML1/ETO, CBFβ/MYH11, TEL/AML1, etc.) in the format of multiplex assays (e.g., Core-Binding-Facor kit) will change routine patient testing. The successful implementation of the BCR/ABL assay in diagnostic workup has been published [146]. The ECOG Leukemia Translational Studies Laboratory has already implemented the BCR/ABL flow cytometry assay in the eligibility screening of leukemia patients for trials that accrue either only BCR/ABLPOS or BCR/ABLNEG patients; in all 70 ALL patients tested, the flow cytometric results were confirmed by subsequent PCR analysis (Paietta E, personal observation). In the case of BCR/ABL, a molecular analysis may want to be run subsequent to the flow cytometric assay, if the BCR/ABL transcript form is of interest. Figure 17.3 demonstrates the clear separation of PE-fluorescence intensities, reflecting the detection of BCR/ABL fusion protein, in a BCR/ABLNEG B-lineage ALL patient, a patient with 10 % BCR/ABLPOS blasts and a patient with 90 % BCR/ABLPOS lymphoblasts. In addition to recording the PE-mean fluorescence intensity and using the negative PE-mean fluorescence intensity range for determining positivity, the ECOG Leukemia Translational Studies Laboratory implemented the intensity ratio for easier comparison between assay runs. This ratio derives from the PE-mean fluorescence intensity of the patient (or positive control) divided by the PE-mean fluorescence intensity of the negative control run in every individual BCR/ABL bead assay. The reason for using this ratio is that the PE-mean fluorescence intensity of negative controls varies ever so slightly among assays (even when using the same cell line) and this variability should be incorporated in the calculation of the extent of positivity of a patient. This approach is analogous to that taken for the calculation of intensities of antigen expression by leukemic cells. While not sufficiently sensitive for MRD, the threshold level of at least 1 % (1 abnormal cell in 100 normal cells) [7] has been found useful in patients who presented with <1 % of circulating lymphoblasts by morphology and flow cytometric immunophenotyping (Paietta E, personal observation).
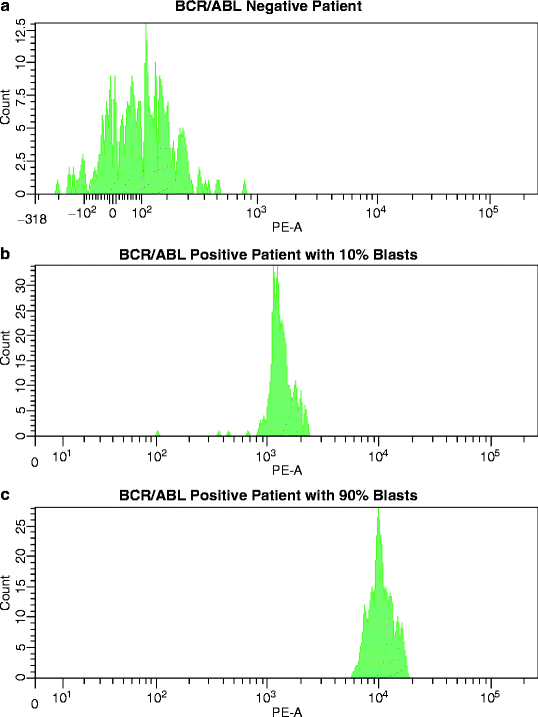
Fig. 17.3
BCR/ABL Flow Assay. Detection of the BCR/ABL fusion protein in mononuclear bone marrow cells from three ALL patients with the BCR/ABL immunobead assay. (a) In this patient, cell lysates failed to yield a PE fluorescence signal greater than the laboratory-established negative mean-value range (mean PE fluorescence 98–190). This patient was confirmed to lack BCR/ABL transcripts by PCR. (b) This histogram shows a mean PE fluorescence signal of approximately 1,500 in a patient with 10 % BCR/ABLPOS lymphoblasts. (c) The bottom histogram represents the strong PE fluorescence signal in a patient with 90 % BCR/ABLPOS lymphoblasts (mean PE signal of approx. 10,000)
Antigens and Therapy
There are several aspects to antigens and antibodies and their usefulness in therapy. First, antibodies to carefully selected antigens expressed by the leukemic cells are administered in in vivo treatment. While purging of stem cell collections with antibodies used to be an important part of this aspect [19], it is no longer of great clinical relevance. Second, therapy may be determined by the specific phenotype of the leukemic cell population. In that case, we speak about phenotype-specific therapy. Both aspects contribute to targeted or personalized therapy in leukemia. Targeted approaches include the use of established surrogate marker profiles for genetic lesions, a subject already discussed earlier in this chapter. The multidrug resistance (MDR) phenotype represents an example of how specific antigens or antigen combinations can reflect functional characteristics of leukemic cells, such as the expulsion of chemotherapeutic drugs. Though MDR modulators have failed to fulfill their promises in the clinic, the strong prognostic association of expression of MDR proteins with outcome, at least in adult AML [147], should keep the interest of scientists alive in elucidating the relationship between MDR and other biologic parameters, such as cytogenetic aberrations. On the other hand, P-glycoprotein (Pgp) expression at initial diagnosis does not appear to be an independent prognostic factor in pediatric AML or ALL [148]. Despite diminishing therapeutic significance at this time, a short description of various MDR proteins and their flow cytometric detection is presented.
Monoclonal Antibody Treatment
In vivo therapy with monoclonal antibodies aims at a specific antigen, which otherwise may be a part of a very diverse immunophenotype. Major requirements for the success of antigen-targeted therapy are the exact knowledge of antigen distribution in normal tissues (cytotoxic effects may be more widespread than intended) and insights into the antigen response to antibody binding (e.g., growth-inhibitory effect of CD33 antibodies is dependent on CD33 phosphorylation and Syk and/or ZAP-70 protein kinase activity) [149]. While there is a plethora of potential antibody targets on leukemic cells, only a select few have emerged as clinically successful [14]. Reasons are multiple and include specificity of antigen expression on target tissue (i.e., the leukemic cell) and the question whether the target antigen is expressed by the LIC in individual patients. Currently, monoclonal antibodies are used mainly in unconjugated form. Alternatively, antibodies can be conjugated and thus function as vehicles carrying immunotoxins or chemotherapeutic agents (e.g., calicheamycin-conjugated CD33 antibody), or conjugated to radioactive molecules delivering selectively to antigen-expressing target cells (e.g., antibodies to CD20, CD22, or CD33 conjugated with various radioemitters, such as [131]Iodine or [91]Yttrium). Radioimmunotherapy with CD33, CD45, and CD164, or CD20 and CD22 antibodies has been tested in AML or ALL, respectively, for its efficacy in intensifying the antileukemic effects of conditioning regimens prior to various types of stem cell transplantation [150, 151]. Targeting two antigens, simultaneously, may yield increased cytotoxicity, as was demonstrated in vitro for gemtuzumab ozogamicin (GO) or Mylotarg, the humanized CD33 monoclonal antibody, and an unconjugated antibody to CD45, a tyrosine phosphatase expressed by the majority of AML and ALL cells [152]. Bispecific antibodies target leukemia-associated antigens while simultaneously activating antigens on cytotoxic effector cells or may otherwise potentiate the signaling events that will eventually lead to inhibition of leukemia cell growth. Bispecific T cell engagers (BiTE) form a new class of constructed antibodies, which direct the body’s cytotoxic T-cells against tumor cells. One BiTE representative is Blinatumomab (MT103), consisting of four immunoglobulin variable domains, of which two form the binding site for CD3 on the surface of T-cells, and the other two form the binding site for CD19 on the surface of the targeted B-cells [153]. Blinatumomab enables a patient’s T-cells to recognize malignant B-lymphocytes and works by linking these two cell types and activating the T-cells to exert cytotoxic activity against the malignant B-cells, causing redirected cell lysis [154]. BiTE antibodies activate T-cells only in the presence of target cells, and nonspecific collateral killing effects have not been observed.
Specificity of antigen expression does not necessarily refer to lineage specificity; for instance, CD33 antibody was successfully added to the treatment of CD33POS myeloid and lymphoid leukemias [14, 155]. Antigen-independent endocytosis has been found to provide a nonspecific uptake mechanism, which presents the rationale for the treatment of both CD33POS and CD33NEG malignancies [156]. The CD33 antigen is not found in nonhematopoietic tissues [19], which makes it an ideal candidate in the use of antibodies for the therapeutic purposes.
Loss of antigen postantibody treatment due to antigen modulation or downregulation causes resistance to immunotherapy. GO, which is linked to the cell toxin calicheamycin, is rapidly internalized after antibody binding, which makes this antigen an ideal candidate for conjugation with intracellularly active cytotoxic agents [157]. The nature of CD33 as an inhibitory receptor [14] sets the stage for its role in cellular interactions and proliferation. CD33 antibody binding induces apoptosis and inhibits proliferation of normal and leukemic myeloid cells. One of the protein kinases involved in the intracellular signaling cascade induced by CD33 antibody ligation is Syk, and the efficacy of CD33 antibody in AML cells has been correlated with the level of Syk expression. Consequently, increasing Syk expression, e.g., with 5-azacytidine, can indirectly enhance CD33 antibody toxicity. Along the same line, the engagement of the SHP-1 tyrosine phosphatase in Syk regulation could be pharmacologically exploited by combining CD33 antibody with cytosine arabinoside and idarubicin [14].
On the other hand, the CD20-antibody rituximab modulates the CD20 antigen by shaving via a mechanism that resembles trogocytosis, the uptake of membrane fragments from one cell by another [158]. The directly cytotoxic effect of rituximab results from various mechanisms of action, including activation of the complement cascade, apoptosis, and recruitment of cytotoxic T cells [159]. There is evidence that the effectiveness of rituximab correlates with the level of CD20 expression by target tissues [160]. With this information in mind, novel CD20 antibodies with higher affinity to the antigen, such as ofatumumab, have been developed, in particular to overcome this potential obstacle in the treatment of chronic lymphocytic leukemia (CLL), a disease characterized by notoriously low CD20 expression [161]. In B-lineage ALL, CD20 expression depends on the maturation stage of malignant B-lymphoblasts both in pediatric and adult patients, but even when expressed, antigen density is quite low [88, 162]. A number of therapeutics have been demonstrated to increase the amounts of surface CD20, for instance, prednisolone [162], while lenalidomide downregulates CD20 levels [163]. Several additional mechanisms of rituximab resistance have been proposed, including the recent discovery of an alternative CD20 transcript, translated into a nonanchored membrane protein, which associates with the CD20 membrane molecule, though, to date, has not been identified in normal B-lymphocytes [164]. Soluble CD20 antigen is another factor affecting the success of antibody therapy, since high levels of circulating soluble antigens reduce the bioavailability and thus efficacy of administered antibody [165].
Another antibody directed at B-lymphocytes is the CD22-antibody epratuzumab. In contrast to rituximab, the antigen–antibody complex is immediately internalized and modulates B-cell activation and signaling [166]. Contrary to CD20, CD22 is equally expressed across all maturation stages of B-lineage ALL [88]. A phase III trial comparing the efficacy of rituximab and epratuzumab in eradicating MRD in adult B-lineage ALL, irrespective of the expression of these antigens, is currently underway in the United Kingdom (A. Fielding, NCRI, personal communication).
CD52 is expressed on virtually all lymphocytes, monocytes, and natural killer cells. In ALL, the density of CD52 antigen expression varies by lymphoid lineage, with T-lymphoblasts demonstrating significantly lower amounts of CD52 on the cell surface [88]. Data with alemtuzumab in ALL are scarce, but suggestive of antileukemic activity [167, 168]. ECOG is currently conducting a phase II study of methotrexate, vincristine, l-asparaginase, and dexamethasone, together with subcutaneous alemtuzumab in relapsed or refactory adult ALL (E1904).
Targeting the Leukemia Stem Cell
The generation of antibodies to leukemia stem cells and their clinical application in patients who achieved a hematological complete remission is an intriguing concept that may be testable, especially in AML. Antigenic features of LIC in AML differ from those of normal stem cells in that they lack CD117, HLA-DR, CD71, and CD90 but express high levels of CD123 [14, 40, 169, 170]. These LICs express valid target antigens [171], such as CD33 [169], CD13, and CD44 [172]. Other antigens that have been shown to distinguish AML LIC from the vast majority of normal stem cells are CD96 [173], CD47 [174], and CLL-1, the C-type lectin-like molecule-1 [175]. Antibodies to CD123, the α-receptor for IL-3, and CD44, a receptor for hyaluronic acid, and CD47, an adhesion molecule, to date, have been tested only in vitro, but have all demonstrated antileukemic activity [14, 40, 170]. On the other hand, the CD34posCD38negCD123pos subpopulation is detectable in very low frequency in normal bone marrows [2, 176].
The high expression of CD33 on the bulk of leukemic myeloblasts and its absence from normal stem cells make CD33 an obvious target for antibody therapy. However, there is evidence suggesting that in some AML subtypes, CD34posCD33neg progenitor cells may be involved, e.g., by monosomy 7 [177] or FLT3-ITD [178] and that involvement of this early precursor population may correlate with poor clinical outcome.
There are exceptions to the concept of a common stem cell phenotype in AML. The LIC in APL, a disease in which the bulk of leukemic cells lacks CD34, has recently been identified as a CD34posCD117pos promyelocyte-like myeloid-committed cell, not a stem cell, in a murine model [179]. Furthermore, in NPM1-mutated AML, a subtype characterized by CD34neg blasts, the phenotype of LIC is heterogenous with LIC found both in CD34pos and CD34neg leukemia-initiating fractions [132].
Many of the antibodies currently used in clinical trials are aimed at antigens not expected to be found on LICs from ALL, for instance, CD20 and CD22 in B-lineage disease [14]. As a side remark, while the majority of multiple myeloma cases lack CD20 on the bulk of abnormal plasma cells, this antigen appears to be expressed by the myeloma stem cell [14]. ALL stem cells are thought to invariably display the cell surface phenotype CD34posCD38neg, though further characteristics remain to be determined [170]. As recently discussed, controversial findings regarding the immunophenotypic profiles of LIC in B-lineage ALL include expression of CD19, CD34, as well as CD38 [180].
MDR Proteins
Flow cytometry can confirm the presence as well as functional integrity of MDR proteins as drug-efflux pumps. The method of sample preparation and the choice of antibodies and of efflux dyes determine the validity of results and conclusions drawn regarding the significance of MDR protein function on clinical outcome.
A recent large randomized, placebo-controlled study of Zosuquidar, a Pgp-modulator without pharmacokinetic interference with chemotherapeutic agents, in elderly AML has revealed some interesting biologic associations [147]. Although Pgp function strongly predicted for poor survival, Zosuquidar had no effect on outcome. The presence of Multidrug Resistance-Related Protein 1 (MRP1) and Breast Cancer Resistance Protein (BCRP) was significantly associated with Pgp, while lung resistance protein (LRP) failed to show a relation to Pgp expression. Greater than 95 % of AML cases expressed two or more MDR modulators on >10 % of myeloblasts. It seems reasonable, therefore, to suggest that the clinical failures of Pgp-modulators, to date, may have been caused by the activities of non-Pgp efflux pumps. Along the line of multifactorial MDR, simultaneous modulation of Pgp and MPR1 was necessary for improved treatment results [181]. Further data suggested that once Pgp and MRP1 were inhibited, BCRP overtook the role of mitoxantrone efflux in vitro [182]. Furthermore, additional MDR proteins, such as MRP family members (MRP2 to MRP5), are found in AML [183]. The relatively higher expression of the various MDR proteins in LICs when compared to normal stem cells [170, 176] supports the notion that the concept of MDR reversal remains a therapeutic strategy of as yet unproven clinical importance. Unfortunately, in the past, MDR has been addressed clinically with naïve simplicity, and the disappointing clinical results with Pgp modulators alone may have discouraged clinical investigators as well as pharmaceutical companies to focus future efforts on this subject.
P-Glycoprotein
The remarkable inconsistencies in the expression and function of Pgp in the acute leukemias may be caused by the use of antibodies to intracellular Pgp epitopes, an unwise choice already discouraged at the First International Workshop on MDR Detection Methods [184], or of antibodies known to exhibit cross-reactivity with non-Pgp molecules [185, 186]. Furthermore, Pgp protein is posttranslationally modified through glycosylation or phosphorylation, the extent of which may differ in various hematopoietic tissues [187, 188], and which can interfere with antibody recognition and Pgp function [187–190]. Even when using an antibody to a surface epitope of Pgp, such as MRK-16, different results must be expected when Pgp is detected on the surface of untreated leukemic blasts, on fixed cells, in thawed cells, or after neuraminidase treatment, to remove glycosylation. This variability in methodology makes it impossible to compare data on Pgp expression across studies.
Other contributing factors are technical pitfalls, such as the use of arbitrary cutoff points of Pgp expression as criteria to define Pgp status [184] and failure to restrict Pgp expression to leukemic blast cells. Substantial Pgp function is found in mature hematopoietic cells, following the order CD56POS natural killer (NK)-cells > CD8POS T-cells > B- and CD4POST cells > monocytes [191–193]. In fact, CD56POS NK cells serve as surrogates in the monitoring of Pgp antagonism in patients receiving MDR modulators in clinical trials [194]. Granulocytes contain MDR1 mRNA (gene encoding Pgp) but lack Pgp protein [193]. Although in normal bone marrow Pgp expression is limited to CD34POS progenitor cells, including myeloid committed precursors [195, 196], normal peripheral blood cells routinely contaminate leukemic marrows and must be excluded by careful flow cytometric gating of the blast cells. The presence of nonleukemic cells presents a problem for MDR1 mRNA analysis because RNA is usually isolated from the total mononuclear cell fraction (consisting of lymphocytes, monocytes, leukemic blasts).
It is important to bear in mind that the detection of MDR protein by antibody staining is not an accurate indicator of its functional activity. Flow cytometric monitoring of rhodamine [123] efflux or accumulation constitutes the standard assay for assessing Pgp function and must be combined with a monoclonal antibody, preferably conjugated with peridinine chlorophyll protein (PerCP) [197], that labels the leukemic cells. Inhibitors of Pgp function in vitro vary in their correlation with Pgp expression in that some modulators produce effects unrelated to Pgp, e.g., verapamil or cyclosporine A, while PSC 833 in combination with rhodamine appears to yield the most sensitive functional Pgp assay [198]. When assaying Pgp function in frozen specimens, cells must be incubated at 37 °C in the presence of fetal bovine serum for at least 1 h after thawing [184, 198]. Prior to performing the functional assay, serum should be removed to avoid any interference with the inhibitory effect of MDR modulators [199].
Effects on Pgp expression data beyond the investigators control relate to polymorphisms of the MDR1 gene, which affect expression and function of Pgp in healthy individuals [200–203]. In AML, variant somatic MDR1 genotypes demonstrated significant associations with MDR1 mRNA expression, cytogenetic risk status, age, and/or response to therapy [204]. Furthermore, age-associated changes in Pgp functional levels were found in bone marrows from normal volunteers, showing continuously increasing function up to the age of 50 years, while remaining constant thereafter [205]. This observation could, in part, explain higher Pgp expression found in elderly AML [147, 206]. On the other hand, the very high Pgp levels reported for patients >58 years [207] may be accounted for by the increased incidence of immature immunophenotypes in those patients [37] and the close association between Pgp function and undifferentiated AML [147, 208]. This association is in line with CD34 being a surrogate marker for classic Pgp drug-efflux activity in AML [209]. In APL, the AML subtype typically lacking CD34 expression on leukemic cells, Pgp is virtually absent [59, 210, 211]. On the other hand, ATRA-resistant APL patients have been found to express Pgp [212]. There exists no such correlation between MDR-1 gene transcription or Pgp function and CD34 expression in ALL [213]. Another antigen associated with Pgp expression is the T-cell affiliated CD7 in AML and in CD4NEGCD8NEG T-lineage ALL, which is thought to originate from the lymphohematopoietic stem cell [214]. Pgp in ALL may be associated with other poor prognostic features, such as high white blood cell count, FAB L3, and a mature B-cell phenotype [215].
The negative prognostic impact of CD56, the neural cell adhesion molecule, in core-binding-factor leukemias [216] is unrelated to Pgp function in this AML subtype [217]. In adult AML, both Pgp function and cytogenetic risk status are powerful predictors of survival [147]. Remarkably, in patients with unfavorable cytogenetics, Pgp status does not further affect survival, while in patients with favorable/intermediate cytogenetics and negligible Pgp function outcome is clearly superior to that in patients from this cytogenetic risk group that have demonstrable Pgp function.
Multidrug Resistance-Related Proteins
Of the six members of the multidrug resistance-related protein (MRP) family, MRP1 and MRP2 are expressed in AML, but only MRP1 has been studied in connection with clinical trials [181]. In a recent large study [147], the presence of MRP1 protein correlated significantly with Pgp expression and the undifferentiated AML phenotype. Controversy exists regarding MRP1 activity in normal CD34pos stem cells [181]. Small technical details that could affect the detection of MRP1 include the use of anti-MRP1 antibody MRPr1 versus MRPm6, which were raised against distinct MRP1 epitopes [218], and variable methodologies for the fixation and permeabilization of the cell membrane which, when tested in parallel, yield strikingly different results (Paietta E, unpublished). Leukemic cells must be surface stained with CD34, CD117, or CD123 prior to fixation/permeabilization to allow gating on the blast cells and avoid contamination with MRP1 expressing normal hematopoietic cells [219, 220].
Functional activity of MRP1 is best measured with calcein AM [221], which does not serve as a substrate for Pgp. The clinical significance of MRP1 in de novo AML remains in question [147, 181, 206, 222]. Increasing evidence points toward the emergence of MRP transporters in relapsed disease [181]. Similar to the findings with Pgp, APL cells lack MRP1 expression [211]. The genes encoding MRP1 and LRP are both located on the short (p) arm of chromosome 16, and one MRP1 allele may be deleted in patients with inv(16)(p13q22) [223].
Little is known about the expression and significance of MRPs in ALL. While MRP1 expression levels were found to be without clinical impact in pediatric ALL [224, 225], MRP3 gene transcription in a single small study was markedly higher in T- than B-lymphoblasts, higher in male than female patients, and associated with poor prognosis [225].
Lung Resistance Protein
LRP expression is highest in AML with monocytic features [147, 206, 226], and in cases without Pgp function [147], the latter being in line with the absence of LRP in APL [211]. The detection of LRP protein by antibody staining faces identical challenges as that of MRP1, given that the specific antibody LRP-56 is directed against an intracellular epitope [227]. LRP expression has not been associated with prognosis in two large cooperative group trials of adult de novo AML of younger (SWOG) [206] and older age (ECOG) [147]. To the contrary, two random analyses of small cohorts of AML patients demonstrated that LRP, but not Pgp, was the most powerful negative prognostic factor for survival [222, 228], highlighting the problematic of underpowered trials. Furthermore, both studies are examples of the detrimental effects of outdated methodology, such as immunocytochemistry, evaluated with arbitrary threshold levels for the definition of LRP positivity in mixtures of normal and abnormal cells. This approach is particularly troublesome as expression of LRP protein has been found in normal bone marrow and peripheral blood leukocytes [229, 230].
Breast Cancer Resistance Protein
Antibodies to BCRP recognize distinct intracellular epitopes [231]; BXP-21 binds preferentially to a functionally inactive state of BCRP, whereas BXP-34 binds only to actively effluxing BCRP [182]. Based on differential binding of these antibodies, Cripe et al. [147] concluded that, at least in elderly AML, the major fraction of BCRP protein at the time of presentation existed in an inactive state. Data on mitoxantrone accumulation have indicated that BCRP effectively transported the drug following the inhibition of Pgp and MRP1 in vitro [182]. Thus, inhibition of one specific transporter may induce activation of BCRP or of other transporters [182]. A similar effect could be expected from failed induction chemotherapy, explaining the increased expression of BCRP in relapsed or refractory AML [232]. Potential interest in BCRP has been sparked by its involvement in imatinib resistance in CML [233]. BCRP activity can be monitored either by using BCRP-preferred substrates or modulators [234].
Antibodies for Predicting and Monitoring Response to Therapy
Therapy in acute leukemia nowadays is predominantly risk-adapted and rarely subtype-oriented. Subtype-oriented therapy applies, for instance, to APL (e.g., ATRA, arsenic trioxide), BCR/ABLpos ALL (tyrosine kinase inhibitors with chemotherapy and allogeneic stem cell transplantation), and core-binding factor leukemias with their exquisite sensitivity to high-dose cytosine arabinoside. Other phenotypes of likely, though yet unproven therapeutic potential are leukemias with FLT3 gene mutations. Several inhibitors of FLT3-tyrosine kinase activity are currently being tested for clinical efficacy in FLT3-mutated AML, with the expectation that patients who share the same molecular defect might respond with equal sensitivity to the same inhibitor. The genetic variability of FLT3 gene mutations, however, impedes any prediction of response [235]. Nonetheless, the success of targeting the FLT3 receptor may be predicted by single cell network profiling (SCNP), which measures the effects of stimulatory or inhibitory therapeutic interventions on multiple signaling pathways [18]. SCNP combines immunophenotyping and potentiated phosphoflow cytometry of cellular responses to discern subpopulations of malignant cells based on their signaling profiles [236]. Assessing the functionality of cellular biochemical pathways in selected cell populations by flow cytometry provides a new tool in the monitoring of molecularly targeted therapy.
Risk-adapted therapy orients itself on established prognostic factors, such as age, white blood cell count, and cytogenetic/molecular aberrations at presentation or level of MRD following induction chemotherapy in morphologically defined complete remission. The powerful negative impact of MRD levels in otherwise standard-risk patients in ALL [15] and AML [16] has prompted increasing efforts to improve and facilitate methodologies for MRD determination. Although a higher sensitivity for detection of remaining leukemic cells is afforded by molecular techniques (e.g., PCR amplification for leukemia transcripts in AML or antigen receptor genes in ALL), flow cytometric methods are currently preferred, because they are applicable in the vast majority of patients, are cheaper, and are easily performed in routine laboratory settings. From scouring the literature, it appears safe to make the statement that although sensitivity levels of molecular and immunologic MRD assessments are not comparable, their prognostic significance appears to be equal. This suggests that below a certain level of residual disease, approximately at 1 abnormal cell in 10,000 normal cells (0.01 %), the remaining disease burden lacks clinical relevance.
Phosphoflow Cytometry
Gene mutations and epigenetic alterations cause leukemic cells to respond to external stimuli in ways different from those seen in nontransformed cells. The method of mapping cellular signaling networks within intact cells using fluorescently labeled phospho-specific antibodies is a rather novel approach, pioneered by Gary Nolan’s group (http://proteomics.stanford.edu/nolan/phospho-flow), to recognize functional heterogeneity among leukemias, beyond standard morphologic, immunophenotypic, cytogenetic, and molecular characteristics, and to monitor the kinetics of response to treatment [18]. This method takes advantage of the power of flow cytometry to interrogate single individual cells, and thus detects cell populations with different signaling properties within a heterogeneous sample. Many proteins involved in cell signaling are phosphorylated. SCNP determines the phosphorylation status of signaling molecules at the single-cell level as a measure of pathway activation or inhibition in cell surface-labeled cells. Phospho-site specific antibodies become increasingly available, enabling the study of signaling events by intracellular flow cytometry (Phosphoflow). By comparing the basal phosphorylation state of proteins with responses to extracellular modulators, the activity levels and kinetics of prespecified signaling pathways can be determined. Phosphoflow cytometry provides insights into cellular signaling in response to therapeutic interventions [18]. Kornblau et al. [237] reported that proteomic profiling with SCNP produced predictive information for response to standard chemotherapy, independent of age and cytogenetics in younger patients with AML. Similar studies are currently underway in AML >60 years old [238]. There is recent evidence that SCNP may provide information beyond that offered by molecular profiling. FLT3 mutations are an example for such situation: To date, only FLT3 receptor mutant AML patients have been selected for trials involving FLT3 receptor kinase inhibitors, with surprisingly variable clinical responses. Downstream targets of FLT3 receptor activation, whether by receptor mutation or FLT3 ligand binding, involve STAT5, PI3-kinase/Akt, and the Ras/Raf/Erk kinase signal transduction pathways which ultimately affect cell survival and proliferation. Functional characterization of those signaling pathways in myeloblasts with mutated versus unmutated FLT3 receptor (FLT3-WT) has revealed heterogeneity among these genetically defined subgroups. In particular, a fraction of FLT3-WT AML signaled as if containing a FLT3 length mutation, while FLT3-ITD patients with low mutation load signaled like FLT3-WT AML (Fig. 17.4) [239].
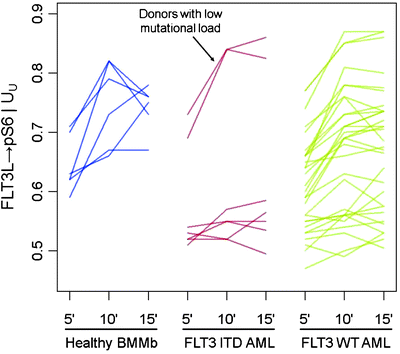
Fig. 17.4
FLT3-ligand induced phosphoflow signaling profiles [239]. Comparison of FLT3-ligand (FLT3L) induced changes in the basal level of phosphorylated S6 (pS6) in healthy bone marrow mononuclear blasts (BMMb), FLT3-mutated (FLT3 ITD), and FLT3 wild type (FLT3 WT) AML blasts, following 5, 10, and 15 min of exposure to FLT3L. Ribosomal protein S6 becomes phosphorylated (pS6) in the MAPK signaling pathway. Compared to BMMb, the majority of FLT3-ITDPOS myeloblasts showed decreased basal levels of pS6 and decreased FLT3L-induced activation of the MAPK pathway, as reflected in pS6 levels. However, two AMLs harboring a low mutant load exhibited a signaling pattern distinct from that of other mutated patients and more similar to BMMb and FLT3 WT cases, possibly suggesting that these patients would respond clinically different to FLT3 kinase inhibition. As expected, the FLT3L-induced response in FLT3 WT patients was widely heterogenous. The Log2(fold) metric on the y-axis is a measure of the fold increase in protein level from the basal state upon modulation. It is the ratio of the median fluorescence intensity of the cell population in the modulated state and basal state on a log scale
Constitutive activation of the phosphoinositide 3-kinase (PI3K)/Akt pathway in de novo AML has been identified as a negative prognostic indicator for overall survival, independent of known prognosticators, such as age, cytogenetic risk group, lactate dehydrogenase, and white blood cell counts at presentation; only the occurrence of FLT3-ITD was marginally inversely correlated with P13K/Akt activation [240]. Another example for the power of phosphoflow is the constitutive activation and phosphorylation of extracellular signal-regulated kinase ½ (p-ERK ½), which is a frequent finding in AML [241]. In ALL, there is evidence that mitogen-activated protein kinase (MAPK) pathway activation may represent an independent prognostic factor for complete remission achievement [242]. There appears to exist no association between p-ERK ½ and age, cytogenetic and molecular findings or immunophenotype, though CD34 expression by lymphoblasts was inversely correlated with p-ERK ½ expression. Mutations of Janus kinases (JAK) have recently been associated with an aggressive type of B-lineage ALL in children, a discovery that relied heavily on intracellular phosphoprotein analysis [243]. At a time of increasing use of tyrosine kinase inhibitor therapy in leukemias, the analysis of the phosphorylation state of oncogenic proteins, such as BCR/ABL, or its downstream effectors, such as STAT5 or CRKL, is essential in assessing the efficacy of kinase inhibition at a given dose and schedule, and its association with clinical response [244]. Treatment of BCR/ABLPOS ALL with an ABL kinase inhibitor leads to the dephosphorylation of the BCR/ABL fusion protein and its downstream effectors, except in cases that carry mutations in the BCR/ABL kinase domain and, therefore, express resistance. Phosphoflow analysis of a blood sample at baseline can provide information on the response to be expected, while samples under treatment could confirm the response. Initial documentations for the feasibility of this approach have been reported [244, 245].
The detection of a small subpopulation of abnormal cells whose signaling profile differed from that of the bulk tumor, providing those cells with a survival advantage during treatment, has allowed for the monitoring of these resistant cells in patients with follicular lymphoma [246]. Consequently, using surface immunophenotyping in combination with phospho-specific flow cytometry for SCNP represents a novel approach to the monitoring of functionally defined MRD.
Minimal Residual Disease Determination by Flow Cytometry
The topic of MRD in leukemia is comprised of several aspects, including (1) the most advantageous methodology for MRD assessment, (2) the selection of peripheral blood versus bone marrow, (3) the timing of MRD assessments, (4) the clinical significance of MRD at the various time points during treatment, and (5) the level of MRD with prognostic power. This section of the chapter focuses on the optimal use of flow cytometry for MRD determination, without entering into any detailed discussions regarding timing issues or clinical relevance.
Leukemia-Associated Immunophenotype
Both in ALL [15, 247–250] and AML [16], pediatric and adult, MRD is a proven independent risk parameter. As a result of the increased implementation of MRD data in treatment stratification, there is dire need for standardization of methodologies. The complex composition of most leukemia antigen profiles facilitates the recognition of low-level disease against the background of normal hematopoiesis. Nowadays, it is accepted practice to establish a LAIP at the time of diagnosis or relapse, or apply a standardized panel of antibody combinations for all MRD cases, in a Different-from-Normal approach [251–253]. A simplified MRD assay applicable exclusively in B-lineage ALL, is based on the hypothesis that CD19POSCD10POSCD34POS/NEG cells detected early after initiation of treatment should be leukemic rather than normal, since normal bone marrow B-cell precursors with this phenotype are exquisitely sensitive to corticosteroids and completely eradicated during remission induction [254]. Before the advent of multicolor flow cytometry, the caveat to keep in mind in the antigen selection for MRD monitoring was the likely appearance of increased normal precursor cells in recovering bone marrows after chemotherapy. B-lymphocyte precursors (hematogones) demonstrate the same sequence of antigen expression as commonly seen in early Pre-B ALL, including CD19, CD10, CD34, and terminal transferase (TdT) expression [255]. Without additional antigenic information, these cells may be mistaken for persistent leukemia [256]. By using four-color flow cytometry at a minimum, a “backbone” set of antibodies, as typified by Coustan-Smith and Campana [15], such as CD19/CD10/CD34, can be combined with a case-specific antibody, e.g., CD33 in ALL. The same concept is applicable to AMLs, in which a “backbone” MRD panel may consist of CD123/CD117/CD33 or CD123/CD34/CD33, etc., combined with CD65(S)/CD15(S) or lymphoid antigens (Fig. 17.5). In T-lineage ALL, the “backbone” should consist of an immature marker, such as CD34, CD117, CD123, a pan-T cell marker, usually CD7 and/or CD5, since CD2 is often missing in T-ALL, and surface CD3, provided that CD3 had been absent from blasts at presentation. Assisting antibodies could be CD1, CD10, CD13/CD33, and a CD4/CD8 combination. Without question, surrogate marker profiles for genetic lesions, as discussed earlier in this chapter, which invariably contain lineage-“foreign” antigens, assist greatly in MRD assessments.
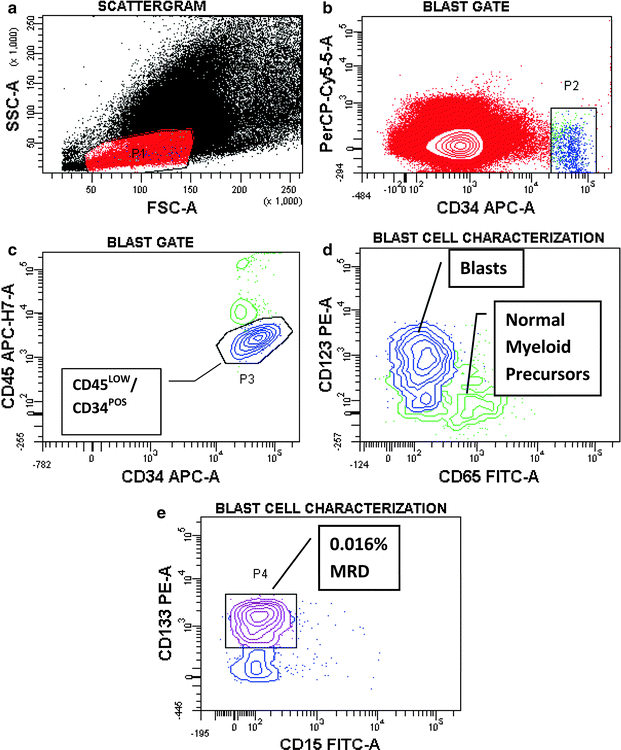
Fig. 17.5
Minimal residual disease (MRD) analysis in a patient with treated AML. Peripheral blood from a patient after treatment for AML was tested and analyzed for MRD by sequential gating for the presence of cells expressing the patient’s leukemia-associated immunophenotype (LAIP). (a) In this scattergram, P1 is drawn around agranular cells (low SSC) of small to intermediate size (FSC) (red dots). (b) In this contour plot, a small population with high CD34 expression is detected and separated from CD34NEG and CD34LOW cells by setting gate P2 (blue dots). (c) By plotting CD34 versus CD45, alleged residual blasts (blue dots) can be further gated away from contaminating normal cells (green dots) still contained within P2. (d) CD45LOW/CD34POS blasts are shown to lack the myeloid antigen CD65, while the contaminating normal myeloid precursor cells express CD65 but lack CD123. (e) Within the CD45LOWCD34POSCD123POS blast population, three-fourth of the cells also express CD133, while lacking CD15, a mature myeloid antigen. These cells account for 0.016 % of white blood cells in this sample and represent MRD. The dense dots in (a) correspond to 4.2 million events (cells) that were analyzed, demonstrating the ability of multiparameter flow cytometry to efficiently characterize very few abnormal cells among huge numbers of normal cells that would otherwise remain undetected
CD34POS precursors in remission bone marrows consist of both lymphoid, generally B-lymphoid, and myeloid progenitor cells. Typically, the B-lymphoid precursors lack myeloid antigens or T-cell markers. Consequently, the finding of myeloid antigens (including CD117) on CD34POSCD19POS CD10POS leukemic B-lymphoblasts provides an excellent tool for MRD monitoring (Fig. 17.6). On the other hand, the normal myeloid CD34POS precursor fraction will express CD117 and early myeloid antigens. Importantly, CD65(S) [257] and CD15 [258, 259], antigens expressed by leukemic myeloblasts in various subtypes of myeloid and/or monocytic leukemias, by lymphoblasts from CD10NEG Pro-B ALL as well as by leukemic cells in rare T-lineage ALLs [3, 4, 19, 257], are absent from normal myeloid bone marrow precursors. Normal CD19POSCD10POS B-cell precursor cells also lack the leukocyte integrin CD11b, which is otherwise found on memory mature B-cells and B-lymphoblasts from all maturation stages of B-lineage ALL and all major genetic subtypes, with the exception of TEL/AML1POS ALL [108]. Rhein et al. [108] reported CD11b not only as a promising novel marker for MRD detection in pediatric B-lineage ALL but demonstrated also its prognostic significance in this disease in that CD11b protein expression at diagnosis correlated inversely with the extent of cytoreduction. Since CD11b expression is present on blast cells of monocytic leukemias and characterizes an AML subgroup with poor prognosis [45], it is important to know that this antigen is not expressed by CD34POSCD117POS normal precursors [257]. A series of phenotypes that might be potentially useful in the monitoring of MRD and that were completely absent from or detectable at low frequency in the blast gate in normal bone marrow samples have been described by Olaru et al. [257].
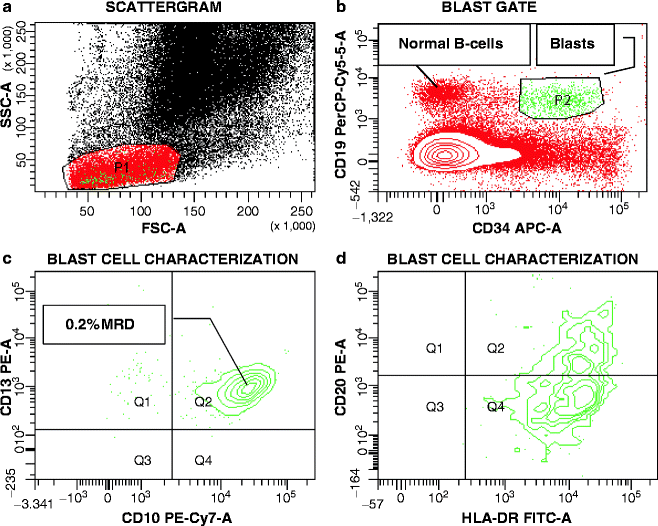
Fig. 17.6
Minimal Residual Disease (MRD) Analysis in a Patient Treated for ALL. Peripheral blood from a patient after treatment for ALL was tested and analyzed for MRD by sequential gating for the presence of cells expressing the patient’s Leukemia-Associated Immunophenotype (LAIP), which consisted of expression of CD34/CD19/CD10, and CD13. (a) In this scattergram, P1 is drawn around agranular cells (low SSC) of small to intermediate size (FSC) (red dots). (b) When cells in the P1 gate are analyzed for CD34 and CD19 expression, two populations of CD19POS cells are identified, CD19POSCD34NEG normal B-cells and CD19POSCD34POS lymphoblasts (green dots). (c) Further analysis of the B-lymphoblasts demonstrates co-expression of CD10 and the myeloid antigen CD13, demonstrating 0.2 % of MRD. (d) As seen in the initial sample, the blasts weakly express CD20
Chen et al. [260] identified a handful of proteins that were expressed in leukemic B-lymphoblasts at higher densities than in normal CD19POSCD10POSCD34POS progenitors, including CD58 (LFA3), a ligand for CD2. Using intensity levels for the distinction of leukemic from normal cells, however, is generally plagued by considerable interpatient and laboratory variability, as exemplified by the limited utility of CD99 in T-lineage ALL [261]. Just as CD99 is predominantly expressed in the thymus in the absence of leukemia, other antigens or antigen combinations present on leukemic cells are confined to healthy tissues other than bone marrow or peripheral blood, for instance TdTPOS lymphoblasts with T-cell markers, CD3, CD5, and/or CD1 [262]. A streamline approach was taken by Krampera et al. [263] in adult T-lineage ALL, in that flow cytometric MRD was determined solely based on the level of cCD3POS/TdTPOS cells among bone marrow mononuclear cells tested during the first year of treatment.
The incidence of suitable LAIPs ranges usually from 75 to 90 % and increases as a function of the complexity and comprehensiveness of the testing antibody panel. The perfect example for this relationship is the published frequency of myeloid antigen expression in ALL. As outlined previously, CD33 and CD13, the myeloid antibodies most frequently tested, are not expressed by the most immature, Pro-B-lineage lymphoblasts, which preferentially express CD65(S) and CD15(S). This explains the extraordinarily high frequency of myeloid antigenPOS ALL cases in ECOG trial E2993, a study that tested seven distinct myeloid antigens. LAIPs most commonly focus on lineage-foreign antigens, such as myeloid antigens in ALL and lymphoid antigens in AML. Nonetheless, some of the custom-designed antigen profiles benefit the most from the absence of antigens, such as lack of CD34, HLA-DR, CD117, CD33, or CD13 expression in AML, of CD10 in B-lineage ALL, and the absence of surface CD3 or any pan-T marker (CD5, CD7, CD2) in T-lineage ALL. Optimally, any combination of MRD “backbone” plus case-specific markers is additionally combined with CD45 antibody to limit MRD evaluation to CD45LOW blast cells. The pan-leukocyte antigen CD45 is expressed by all myeloid, monocytic, and T-lymphoid leukemias, but can be absent in a fraction of B-lineage ALL. The intensity of CD45 staining of blast cells is characteristically lower than that of mature hematopoietic cells contaminating a test tissue, with the exception of granulocytes, which demonstrate weak CD45 expression. Consequently, the first step in the characterization of blast cells is the gating of cells with low side scatter, away from granulocytes and monocytes, prior to gating on CD45LOW cells (Fig. 17.7). The CD45 APC-H7 conjugate is an excellent tool both during initial evaluation and MRD assessment. APC-H7 is a new APC-cyanine tandem dye, which has a fluorescent emission maximum of 767 nm, away from all of the commonly used fluorochromes. A CD45-H7 versus side-scatter display is also of invaluable help if MRD studies are to be performed without having been able to test a patient’s initial leukemia population. In that situation, a “Pretest,” as outlined next, run on CD45LOW cells will offer the best chance to identify any blasts present.
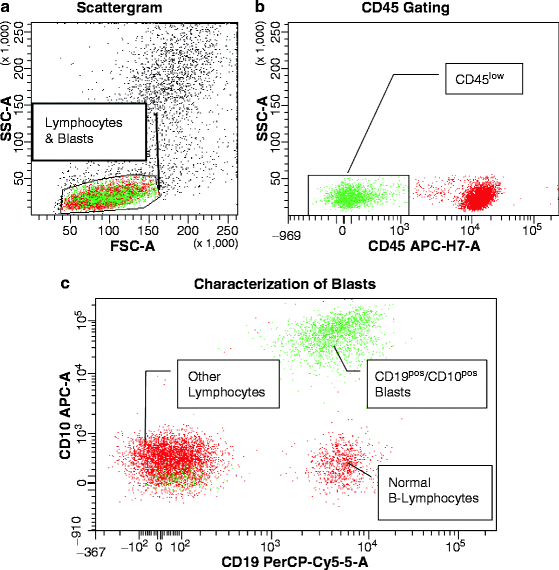
Fig. 17.7
Example of the usefulness of CD45 conjugated to APC-H7 in the selection of leukemic blast cells in a patient with CD19/CD10POS B-lineage ALL by sequential gating. (a) In this scattergram, peripheral blood lymphocytes and blast cells are gated upon based on their small to intermediate size (FSC-A) and low degree of granularity (SSC-A). (b) When analyzed for CD45 expression, blasts with their characteristically low CD45 staining intensity (green dots) are separated from normal lymphocytes (red dots). (c) Only the CD45LOW lymphoblasts (green dots) show double expression of CD19 and CD10, diagnostic of B-lineage ALL
Without question, flow cytometric MRD would be helped immensely by the availability of suitable antibodies to leukemia-specific fusion or mutant proteins. The recently developed flow cytometric kit for BCR/ABL proteins has demonstrated a sensitivity threshold of 1 %, though it did reach 0.1 % with some cell lines [7]. Although this level is 1–2 logs higher than that to be reached for true MRD measurements, continued work on improved methodology holds promise to make these assays applicable to MRD studies, in particular with better control of protease activity [72]. Currently, specifics are not yet known regarding the sensitivity levels of antibodies to mutated NPM1 protein [144, 145].
Access to a flow cytometric assay for PML/RARα proteins, as promised for the near future [72], will clearly simplify MRD detection in APL. Although PML/RARαPOS APL displays a distinct immunophenotype [31, 33], this characteristic antigen pattern offers little with respect to MRD detection, and the variable light scatter characteristics of leukemic promyelocytes [31, 33] complicate matters further. However, a number of antigenic features are useful: (1) the expression of the T-cell affiliated antigen CD2 by leukemic promyelocytes carrying the S-isoform of PML/RARα allows for localizing residual disease—care has to be taken to check the intensity of CD2 expression on initial APL cells, since it could have been quite weak and should not be expected to be stronger posttreatment; (2) the lack of CD34 and/or HLA-DR despite CD117 expression, albeit typically weak; (3) the lack of CD11a/CD18 expression on otherwise normal appearing myeloid cells serves as indicators for MRD; and (4) finally, and most importantly, leukemic promyelocytes express the sialylated form of CD15, while on normal promyelocytes asialo-CD15 is found; unfortunately, the reactivity patterns of CD15 antibodies with these two forms of the antigens are usually poorly described in commercial data sheets; for laboratories truly interested in assessing the binding specificity of their CD15 antibodies(s), it is recommended to test cell lines known to express the sialylated CD15 form (e.g., neuroblastoma cell lines, e.g., SK-N-SH, SK-N-MC) and re-resting them after neuraminidase treatment.
Pretest Panels
For every new case of acute leukemia, the primary question to be answered is that of lineage affiliation. At the same time, APL should be excluded as quickly as possible. To accomplish these goals, a limited antibody panel can be run, which we have denoted the “Pretest” panel (Table 17.3). If any question remains with regard to the cell lineage, myeloperoxidase in combination with cCD22 or cCD3 needs to be assayed on the blast cells exclusively prior to continuing the testing. Once the cell lineage has been confirmed, and APL has been ruled out, predominantly based on CD11a/CD18 negativity in a myeloid phenotype that lacks CD19 and/or CD56 (features of AML1/ETOPOS AML, one other disease subtype that lacks the integrins), as outlined previously, additional markers can be tested. Their choice will depend on whether supplementary antibodies are necessary to substantiate a suspicion of megakaryocytic or erythroid leukemia (e.g., CD41, CD42, CD36, myeloperoxidase), whether immunotherapy is planned requiring, for instance, CD52 or CD20 determination, whether secondary antigens need to be assessed to evaluate the level of maturation, particularly in ALL (e.g., CD4/CD8, CD62L, CD57 and TCR proteins in T-lineage ALL, surface and cytoplasmic IgM, surface immunoglobulin light chains and CD20 in B-lineage ALL, or TdT in both), or whether ancillary antigens are sought in an attempt to establish the optimal LAIP for future MRD determinations. These reasons are not mutually exclusive and, usually, one subtype-specific panel will fulfill all requirements, sometimes with slight adjustments.
Table 17.3
Acute leukemia pretest
TUBE # | Fluorochromes | |||||
|---|---|---|---|---|---|---|
FITC | PE | PerCP-Cy5.5 | PE-Cy7 | APC | APC-H7 | |
1 | CD45 | CD14 | CD123 | CD34 | ||
2 | CD65 | CD2 | CD3 | CD2 | CD5 | CD45 |
3 | CD11a | CD56 | HLA-DR | CD117 | CD45 | |
4 | CD18 | CD133 | CD19 | CD13 | CD10 | CD45 |
5 | CD7 | CD24 | CD41 | CD33 | CD11b | CD45 |
MRD Levels in Peripheral Blood Versus Bone Marrow
The question as to whether peripheral blood qualifies as a source for MRD determination is an important one given the ease of blood drawing compared to the discomfort of bone marrow aspiration. This subject has been addressed extensively for the molecular MRD monitoring of BCR/ABL in CML [264]. In BCR/ABLPOS ALL, residual BCR/ABL levels in blood specimens were significantly lower than those measured in bone marrow aspirates from the same time point [83]. With respect to immunologic MRD, comparative analyses in ALL have shown beyond doubt that the two tissues are equivalent in T-lineage but not in B-lineage ALL, whether analyzed by RT-PCR [265] or flow cytometry [266]. In a large fraction of B-ALL patients, blood samples were negative for MRD by either methodology when the bone marrow was still positive. Despite these discouraging results, Coustan-Smith et al. [266] reported that those cases of B-lineage ALL that had positive corresponding blood samples were at very high risk of disease recurrence, implying that the testing of both tissues, not only of bone marrow, might be clinically important in this disease. With respect to AML, MRD examinations have focused on bone marrow aspirates. However, data of flow cytometric MRD monitoring in the recently closed ECOG phase III trial, E1900 [27], suggest that immunologic MRD levels are consistently lower in the blood than the bone marrow at all time points examined (Paietta, unpublished observation). The value of assessing MRD in peripheral blood not only depends on disease subtype but also the reason for testing. If the purpose of measuring MRD is to determine the rate of blast clearance following induction chemotherapy, serial blood samples will do the job. If, however, absolute MRD levels are to be determined at the time of complete hematologic remission, a bone marrow aspirate will be required. As yet undetermined is the effect of growth factor treatments on the level of circulating blasts; it seems wise, therefore, to perform MRD measurements when patients have completed their treatments and have fully recovered their blood counts.
Stability of Leukemia-Associated Immunophenotypes with Treatment
The primary limitation of immunologic MRD measurements in the literature remains to be a potential change in the selected LAIP with treatment, including, in extreme cases, even a lineage switch. Reports on profound phenotypic alterations exist predominantly in the older literature [19, 262]. Several more recent studies have continued to use suboptimal methodology, such as improper antibody and fluorochrome combinations, and thresholds for the definition of antigen positivity that were unacceptably low (e.g., 10 %) given that gating strategies for blast cells did not unequivocally exclude contamination by normal cells [267, 268]. In other cases [269], LAIPs detected overwhelmingly at relapse were clearly present at diagnosis, though only in a small subset of the leukemic bulk population. To call this occurrence a phenotype switch is not justified and simply reflects the necessity to carefully evaluate presentation immunophenotypes for multiple LAIPs [16]. The assumption that antigens expressed weakly at the initial blast cells may have a stronger tendency to be lost after treatment or at relapse [19] may have also derived from inexperienced interpretation of antigen expression patterns and/or incomplete control of nonspecific binding at initial testing.
Although multiple LAIPs can exist in individual patients at the time of diagnosis, in most cases, these phenotypes most likely represent a continuum of differentiation stages of one underlying clone. This notion is analogous to clonal karyotypic evolution, and, in fact, it would not be surprising to find antigenic differences to be aligned with cytogenetic subclones. Not unexpectedly, some LAIPs are lost [16] and overall antigenic modifications occur among LAIPs with treatment, such as the observed trend towards de-differentiation, reflected, for instance, in a loss of CD10 in ALL or of CD15 in AML [16, 19]. On the other hand, Gaipa et al. [270] found the downregulation of CD10 and CD34, while CD19, CD11a, and CD20 were upregulated during the initial phase of induction treatment for pediatric B-lineage ALL, indicative of progressive maturation. Remarkably, normal B-lymphocytes present in the specimens were equally affected. In support of the theory that prednisone therapy affects antigenic gene transcription, the same investigators subsequently reported that these changes in antigen expression, except for CD11a, reverted to initial levels later during treatment [271]. Rhein et al. [108] found CD11b to be consistently and increasingly upregulated in leukemic B-lymphoblasts that persisted after therapy. This group had previously reported that gene expression during induction therapy in B-ALL shifted towards that of normal B-cells [272]. In ECOG’s experience, certain antigenic features that constitute the strongest part of any LAIP persist during treatment and reappear at relapse. These features are antigens that represent surrogate marker profiles for genetic lesions, such as CD19 in AML1/ETOPOS AML, CD2 in CBFβ/MYH11POS AML or APL, CD33 and CD13 in BCR/ABLPOS ALL; furthermore, in AML, lack of CD34 or CD117 and expression of CD7 and CD11b are very stable. On the other hand, expression of CD64, CD41, and CD133 is variable (Paietta E, unpublished).
To ensure that antigenic modulation does not interfere with MRD monitoring, it is recommended to combine as many leukemia-characteristic antigenic features as possible in the LAIP of choice so that loss of one or even two distinguishing attributes will not hamper the monitoring of MRD.
Lineage-Specific Antigens
In at least 95 % of acute leukemias, a single dominant lineage affiliation can be established through the detection of one of three lineage-specific antigens, myeloperoxidase (protein, not function), cCD22 or cCD3, for the myeloid, B- or T-lineage, respectively. The proposed specificity of cytoplasmic CD79a for B-cell-related ALL and potential pitfalls with its usefulness have been previously discussed in detail [19]. It is essential that myeloperoxidase be tested simultaneously with cCD3 or cCD22 in leukemic blasts that are gated flow cytometrically either through side-scatter versus CD45 or through a gating antibody, such as CD34 or CD117. Leukemia populations with truly “biphenotypic” features are rare and manifest themselves through dual expression of two lineage-specific antigens in the same cell, e.g., myeloperoxidase and cCD3 as seen in some cases with CD117POS ALL [11]. An example of a biphenotypic subpopulation with dual myeloperoxidase/cCD22 expression in a case of otherwise B-lineage ALL is shown in Fig. 17.8. It is important to remember that CD22 and CD3 can both be found on the surface of mature B- and T-lymphocytes, respectively; control staining of the cell surface and gating exclusively on abnormal cells are an absolute prerequisite for meaningful results. The intracellular localization of these crucial lineage-specific antigens is unfortunate because their detection by flow cytometry requires technical skill and experience. In fact, routine clinical laboratories may shy away from testing intracellular antigens by flow cytometry. Aside from choosing optimal fixation and permeabilization media, it is essential that data on intracellular antigens reflect gated blast cells. Following fixation and permeabilization, scattergram characteristics (size and granularity) of cell populations change, often leading to the collapse of populations that could be easily distinguished in unfixed specimens. This situation creates a problem, especially if blast cells lack immature antigens, which are routinely used to separate leukemic from normal cells (e.g., CD34, CD123). As an example, CD5 and CD3 antibodies are useful in the detection of surface CD5posCD3neg T-lymphoblasts and separation from normal surface CD5posCD3pos T-lymphocytes. Thus, both of these antibodies must be combined with the myeloperoxidase antibody to restrict detection of antigen in T-lymphoblasts. If similar measures are not employed, it can lead to misleading reports, such as the finding of myeloperoxidase protein expression in a high percentage of ALLs.