Fig. 32.1
Dose-toxicity and dose-efficacy relationships with immune checkpoint blockers. Contrary to cytotoxic agents (left panel), immune checkpoint blockers (right panel) do not display any linear dose-toxicity or dose-efficacy relationship. Rare, but severe and sometimes life-threatening, immune-related adverse events (irAEs) can be observed at all dose levels; biological activity is independent of the dose above a certain threshold required for triggering the immune response. The therapeutic index of these therapies is consequently wider than the one of traditional cytotoxic agents. Color code: mild, moderate, and severe toxicities are represented respectively with green, orange, and red lines; red dots represent the potential for severe irAEs on a backbone of dose-independent mild AEs with immune therapies; activity is depicted in yellow. DL dose level, MAD minimum active dose, MFD maximum feasible dose, MTD maximum tolerated dose, RP2D recommended phase 2 dose, Ther Index therapeutic index
32.2.1.1 Safety and MTD Definition
Toxicities of immune therapies differ in several ways from toxicities observed with cytotoxic therapies or targeted agents. If a dose-toxicity relationship has been observed with anti-CTLA-4 agents, no correlation between dose and safety profile has been observed in phase 1 trials evaluating anti-PD-1 or anti-PD-L1 agents as monotherapy—which was confirmed in later phase studies. For example, nivolumab (Opdivo®, Bristol-Myers Squibb) efficacy and safety profile appears relatively constant at doses ranging between 2 and 20 mg/kg either Q2W or Q3W [3–5]. Also, when considering the 13 main phase 1 trials evaluating anti-CTLA-4, anti-PD-1, or anti-PD-L1 agents as monotherapy, only one trial identified some per-protocol-defined DLTs, and no MTD could be determined in almost all trials. Therefore, the recommended phase 2 dose (RP2D) was most frequently established as the maximum feasible dose (MFD)—rather than being based on safety data. Interestingly, the nivolumab recommended dose was established based on an integrated analysis of pharmacokinetic (PK, reflecting drug exposure), pharmacodynamic (PD), and safety and efficacy data of a large phase 1b study [3, 6]. Similarly to targeted therapies, most immune checkpoint blockers are eventually administered at fixed doses, which are neither adjusted on body weight, body surface area, nor any patient-specific parameter (Fig. 32.1).
Does this absence of dose-limiting toxicities or MTD mean that these therapies are not toxic? If the number of severe toxicities observed with immune checkpoint blockers is indeed generally lower than with cytotoxic therapies or targeted therapies, this absence of DLTs is mainly due to a difference in kinetics of toxicity appearance. DLTs are traditionally assessed during the first cycle of therapy only, which is relevant for toxicities that are directly resulting from an acute effect of the drug on healthy cells. By contrast, toxicities resulting from immune therapies are mostly indirect: they are caused by an inadequate response of the immune system to the drug—which can be inadequate in nature or in quantity—and inappropriate reprograming of immune cells. This has led to the distinction between adverse events (AEs) and immune-related adverse events (irAEs)—including infusion reactions. A latency period, which duration varies according to the nature of the drug (e.g., 8–10 weeks for ipilimumab [7]), is necessary before irAEs can occur. Therefore, a DLT period limited to the first cycle only does not appropriately capture such drug-related toxicities, and these are not taken into account in the dose-escalation process. If the latter is completely acceptable, considering the frequent absence of dose-toxicity relationship as well as necessity to complete phase 1 trials in a timely manner, such late toxicities should however be carefully reported in phase 1 manuscripts together with their cycle of occurrence. Alternatively, some phase 1 trials assessing anti-PD-L1 agents, such as trials evaluating BMS-936559 [8] and MEDI4736 (durvalumab, AstraZeneca) [9], have made the choice to lengthen the DLT period to two cycles, in order to better take immune-related toxicities into account.
Dose-limiting toxicities may also not be limited to severe (NCI-CTCAE Grade 3) toxicities in phase 1 trials evaluating immune therapies. Indeed, irAEs require rapid and specific management—based mostly on immunosuppressive and anti-inflammatory agents such as steroids, anti-TNF, or anti-IL-6 agents in more severe cases. Therefore, most trials now recommend discontinuing drug administration as soon as moderate (Grade 2) irAEs are observed and starting rapid therapeutic intervention according to dedicated guidelines [10]. Even if some uncertainties remain regarding the pharmacokinetics of elimination of immune checkpoint blockers from their target, drug interruptions are likely leading to decreased drug exposure and should therefore score as DLTs according to the most recent recommendations [11]. Most importantly, such delayed toxicities should deserve specific attention and should be taken into account in the dose-recommendation process as well as in the establishment of guidelines for toxicity management [12].
32.2.1.2 Dosing Schedule
In most cases, immune therapies do not present a clear and linear dose-response relationship (Fig. 32.1). For example, the activity has consistently been observed at all dose levels in the phase 1 trial evaluating several doses of nivolumab [3]; similarly, higher doses of the anti-CTLA-4 agents ipilimumab and tremelimumab did not result into higher response rates in melanoma patients. For example, the dose-response suggested in the phase 2 trial comparing three doses of ipilimumab in metastatic melanoma [13] was not confirmed in subsequent larger phase 3 trials [14–16]. Therefore, aiming at administering the highest tolerable dose might not be relevant for these agents, and their potential for efficacy at lower dose levels should be explored. In this context, innovative designs, allowing to dose-escalate rapidly (accelerated titration designs or modified toxicity probability interval designs [17]) and to expand at low dose levels as soon as satisfactory pharmacokinetic and pharmacodynamic parameters have been obtained, should systematically be considered (Fig. 32.2).
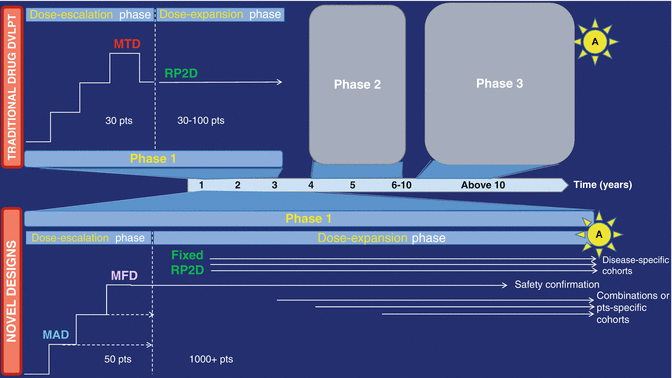
Fig. 32.2
Novel phase 1 designs in the era of immuno-oncology. The traditional drug development process (top panel) includes three separate phase 1, 2, and 3 studies prior to reaching drug approval (depicted by “A” in the sun shape). Classical phase 1 associates a dose-escalation phase followed by a single-cohort dose-expansion phase primarily looking for safety and confirmation of the recommended phase 2 dose. This approximately 3-year-long phase 1 is, in case of data supporting preliminary activity, followed by subsequent phase 2 and 3 trial, for a total drug development duration of more than 10 years. Novel drug development designs include “drug registration phase 1 trials,” in which expansion cohorts do not only look for safety but also activity in disease-specific cohorts that are adequately powered to do so. Further, multiple combination cohorts can be launched as a part of the original phase 1 trial (pending appropriate protocol amendment), to assess safety and preliminary efficacy of various drug associations. These multiple parallel cohorts can include several hundreds of patients and lead to breakthrough designation, conditional, or accelerated approval, thereby significantly shortening the drug development process. Patient-specific cohorts may also be open, in order to allow assessing the drug’s safety in patients presenting certain comorbidities. Additional patients could also be enrolled in the dose-escalation phase at dose levels which have proven to be safe and are above the minimum active dose; this allows enriching for safety, PK, PD, and efficacy data at several dose levels while increasing the number of patients who will potentially benefit from the drug, without any delay in dose-escalation. MAD minimum active dose, MFD maximum feasible dose, MTD maximum tolerated dose, RP2D recommended phase 2 dose
Beyond the question of the dose, the optimal schedule of administration also remains debated. If most monoclonal antibodies are currently administered every 2 or 3 weeks, no consistent pattern can be found according to the antibody isotype, and there are still several unknown parameters—such as antibody clearance from its target—which require further exploration. Phase 1 trials should endeavor to best explore such questions, as detailed below (see pharmacokinetic and pharmacodynamic parameters section). The total duration of treatment is also the matter of intense debate. For example, if anti-PD-1 and anti-PD-L1 agents are currently administered following a continuous schedule as monotherapy, the anti-CTLA-4 agent ipilimumab is currently approved at 3 mg/kg Q3W for a total of four doses, and some studies evaluated the potential for maintenance therapy with dosing at lower frequency [18]. The potential for effective reinduction therapy for patients who present sustained tumor response off-treatment but ultimately relapse should also systematically be considered [19]. However, if phase 1 trials are the place for testing the feasibility and safety of such schedules, the formal comparison of their efficacy requires on-purpose adequately powered later phase trials in dedicated tumor types.
32.2.2 Safety Profile and Toxicity Management
A full chapter of this book being dedicated to side effects of I-Os, only key notions and historical developments of interest for phase 1 trials are presented here.
There are several specificities of immune toxicities (immune-related adverse events, irAEs) that need to be considered in phase 1 trials (Fig. 32.3). First, irAEs can affect any organ, even if initial trials with anti-CTLA-4 and anti-PD-1/PD-L1 agents mostly focused on colitis and pneumonitis [10, 20]. Any irAE semiology should therefore be carefully and exhaustively described in the medical records. Second, their potential for rapid worsening and life-threatening consequences contrasts with their low incidence. Therefore, patients should be systematically taught to promptly report any symptom without delay and avoid self-management. Third, as irAEs require specific management, phase 1 protocols should implement clear toxicity management guidelines, and patients should be referred to the local organ specialist as soon as irAE reaches Grade 2 [10]. Phase 1 units should ideally set up a collaborative network with organ specialists in order to optimally explore and manage irAEs. Beyond optimizing treatment, this would allow collecting tissue or blood samples early—while the patient still experiences the toxicity and before any immunosuppressive agent has been administered—in order to study its physiopathology.
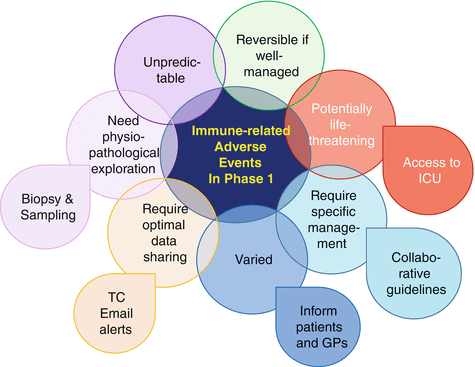
Fig. 32.3
Specificities of irAEs requiring peculiar attention in phase 1 trials. GP general practitioner, ICU intensive care unit, TC teleconference
If antibodies targeting immune checkpoints are currently seen as “safe nontoxic” drugs, early developments have proven to be sometimes hectic. The most famous example resides in the story of the CD28 superagonist TGN1412, developed in 2006 by TeGenero [21]. All six healthy volunteers who received the first drug injection developed life-threatening cytokine release syndromes requiring transfer to intensive care units within a few hours of the drug administration. Although all six survived, this event put the development of therapies targeting the immune synapse in the shade for several years. The causes of such severe toxicities were eventually deciphered [22] and included the inadequate choice of the starting dose—which should have been based on the minimum biological effect level (MABEL) instead of the traditional non-observed adverse effect level (NOAEL), as well as differences between the immune systems of humans and animals which had been passed over (including differences in memory T cells in mice and macaques). This example, associated to others that will be discussed below with vaccines and CAR-T cell therapies, illustrates how toxicities of I-O agents are unpredictable and, even if rare, are often severe and require prompt and specific management in specialized units. Therefore, phase 1 trials of immune therapies should, at least for first-in-class or first-in-human drugs, solely be performed in large experienced phase 1 centers with immediate access to intensive care unit and efficient immunosuppressive agents (including anti-TNF and anti-IL-6 agents).
32.2.3 Pharmacokinetic and Pharmacodynamic Assessments
32.2.3.1 Pharmacokinetics
Considering the mechanism of action of immune therapies and the variety of agents currently assessed in phase 1 trials, pharmacokinetic evaluation is becoming increasingly complex. Mechanism of absorption, distribution, metabolism, and elimination is highly variable according to the type of agent [23]. For example, if most immune checkpoint blockers have a common IgG backbone—which accounts for a dose-dependent C max and a median half-life of approximately 15 days—differences exist according to the isotype. IgG1 and IgG3 can cause NK cell-mediated antibody-dependent cell-mediated cytotoxicity (ADCC), whereas IgG4 rather activates the alternative complement pathway. Other factors, such as a concentration-dependent half-life, the ability of the Fc region to bind to the salvage receptor (FcRn) [24], or the presence of circulating soluble forms of the ligand, introduce further variability and complexity in the assessment of pharmacokinetic characteristics of these agents. By contrast, bi-specific antibodies lacking an Fc region traditionally have a very short half-life, which can be of interest notably for the management of drug-related adverse events, such as cytokine release syndrome or appearance of anti-drug antibodies (ADAs), either human anti-human antibodies (HAHAs) or human anti-mouse antibodies (HAMAs) [25, 26]. The bi-specific T-cell engaging (BiTE®) antibody anti-CD19xanti-CD3 blinatumomab (Blincyto®), for example, has a half-life of approximately 2 h [27]. Finally, novel routes of administration, such as intra-tumor administration, lead to novel challenges, with a maximum administrable volume or the requirement to—ideally—assess the drug’s pharmacokinetics not only in the peripheral blood but also locally. The optimization of the drug formulation is therefore increasingly important.
32.2.3.2 Pharmacodynamics
The difficulty in designing appropriate pharmacodynamic assays for immune checkpoint blockers resides in their highly variable level and pattern of expression. For example, PD-1 is mostly expressed on T-cell surface, while CTLA-4 only displays transient expression, and biomarker expression can both be constitutive or inducible [28]. Thus, flow cytometry methods assessing receptor occupancy on circulating T cells are feasible and relevant only for a fraction of molecules. Such technique could successfully be used for the phase 1 trial evaluating nivolumab [29], where receptor occupancy appeared to be dose-independent and prolonged (with a decay in occupancy being observed only 3 months after the last drug administration). Importantly, which degree and duration of target modulation should be achieved to trigger the optimal immune modulation, and whether findings observed in circulating lymphocytes reflect what is happening in the tumor bed, remains unknown. Further, data have highlighted the critical role of the interaction of the Fc portion of monoclonal antibodies with FcγRs expressed on innate immune effector cells in therapeutic activity. Patients displaying activating FcγR alleles (resulting in higher affinity) respond better to therapy, and polymorphisms in the FcγRIIIα were associated with response to rituximab in patients with follicular lymphoma [30, 31]. Therefore, strategies have been developed to limit or abrogate the binding of the antibody Fc to cellular FcγRs, which may introduce further variability in drug efficacy and pharmacodynamic parameters [32].
The ability to perform immunomonitoring studies, i.e., assessing the levels of circulating cytokines, chemokines, as well as phenotyping immune cell populations in peripheral blood, represents an attractive pharmacodynamic biomarker for immune therapies. For example, the phase 1 trial assessing the 9B12 anti-OX40 monoclonal antibody evidenced that, following drug infusion, patients displayed a non-Treg (FoxP3neg) CD4+ T cell expansion associated with an increase of CD8+ lymphocytes expressing the CD38 and HLA-DR activation markers, together with a transient decrease of follicular helper CD4+ T cells in the peripheral blood [33]. The comparison of OX40 surface expression on Treg from the peripheral blood and on tumor infiltrating Tregs in three patients for which tumor tissue was available revealed that less than 20% of circulating Treg expressed this marker, whereas more than 50% of infiltrating Tregs were OX40 positive. By contrast, anti-CTLA-4 has been reported to increase both FoxP3+ and FoxP3− CD4+ T cells in a dose-dependent manner.
Such extensive and comprehensive pharmacodynamic studies are essential as early as phase 1 trials, even if such studies are not powered to show any statistically significant differences in biomarkers. Indeed, phase 1 trials are the only situation where the effect of the dose schedule and drug exposure on PD parameters will be evaluable. Further, this will allow orientating biomarkers that should be evaluated as a priority in later phase trials that will enroll a larger number of patients, thereby limiting the cost of such analyses.
32.2.4 Patient Eligibility
Considering the peculiar physiopathology of the immune therapy-associated toxicities, and the “phase 1 efficacy and registration trial” development strategy currently chosen by most drug companies (Fig. 32.2), one can wonder whether the traditional phase 1 eligibility criteria should be unwound. Patients included in phase 1 trials are usually required to have (1) life expectancy above 18 weeks and (2) no major baseline organ dysfunction. Life expectancy has traditionally been assessed using objective scores developed on chemotherapy-treated patients, such as the Royal Marsden score based on LDH, albumin, and number of metastatic sites [34]. Recent data have revalidated the relevance of such scores on immune therapy-treated patients and highlighted the potential for adding lymphocyte count in the scoring [35]. Regarding the criteria excluding patients with any baseline organ dysfunction, phase 1 trials evaluating immune therapies represent a major paradox. Indeed, recent data of antibodies targeting immune checkpoints tend to support that immune-related toxicities might be much rarer and varied than chemotherapy- or targeted agent-associated toxicities [10]. However, inclusion criteria of phase 1 trials have become more restrictive. Logically, patients with history of autoimmune disease (psoriasis, type 1 diabetes, inflammatory bowel disease, etc.), chronic viral infection (hepatitis B, hepatitis C, HIV), or history of severe allergic, anaphylactic, or hypersensitivity reactions have been excluded from such trials. However, if such narrowing of the inclusion criteria is legitimate at the very first development steps of a novel drug, i.e., during the dose-escalation phase or for first-in-class/first-in-human compounds, it further decreases the representativeness of phase 1 patients of the all-comer populations. This is contra-intuitive in a context where drug companies aim at obtaining accelerated approval for some compounds based on phase 1 trials results. Responses and clinical benefit have been observed in patients with elevated LDH receiving tremelimumab [36], in patients with performance status of two treated with BMS-936559 [8], or in patients with brain metastases [37, 38] with no additional toxicity. Provisions should therefore be made to open dedicated cohorts of patients with poorer prognosis in the dose-expansion phase, after unwinding of selected eligibility criteria, chosen based on the drug safety profile that has been observed in the dose-escalation phase and safety dose-expansion cohort. Also, evaluating such “more at risk” patients within the initial phase 1 trial would allow that physicians looking after them are already familiar with the drug, which is ethical and clinically sound. Together with the removal of some limitations put on the number of previous treatment lines, this would allow speeding up recruitment and expediting drug development, while obtaining additional safety information on the evaluated compound. Finally, most phase 1 trials currently require patients being immune therapy-naïve. With the exponential development of immune therapies, this becomes no longer feasible as most patients will have received at least one prior line with an immune modulator. Unless there is a strong rationale and fear of interaction that would put the patient’s safety at risk, such criteria should be removed.
32.2.5 Patient Selection and Personalized Immune Therapy
Immune therapies have brought an additional level of complexity in the patient selection and personalized medicine era. Biomarkers are not anymore located only on cancer cells (or molecules released by cancer cells), but immune cells and tumor microenvironment need to be taken into account [39]. Further, some biomarkers—such as PD-1 or PD-L1—have a dynamic expression profile and can be induced under specific circumstances only [28]. The challenges of choosing the best biomarker for predicting efficacy to immune therapies—including checkpoint expression, neoantigen/mutational load, immunoscore, etc.—have been discussed in a previous chapter of this book, but several phase 1 specificities need to be highlighted. First, phase 1 trials need to remain limited in time and able to enroll patients efficiently. Therefore, no molecular selection should be performed in the dose-escalation phase, where the primary objective must remain the determination of the dose-limiting toxicities and maximum tolerated dose. Molecular enrichment could then be performed in the dose-expansion phase in dedicated “companion biomarker” cohorts. The successful development and approval of the 22C3 IHC PD-L1 PharmDx test, together with pembrolizumab, nicely illustrates this concept [40]. Second, none of the current biomarkers used to predict response to immune therapies, notably immune checkpoint blockers, is sufficiently specific or sensitive to exclude patients from receiving such therapies. For example, responses to anti-PD-1 or PD-L1 agents have consistently been observed in PD-L1-negative populations [41, 42] or in tumor types with virtually no mutations and a very low neoantigen load—such as Hodgkin lymphomas [43]. Such observations would have been missed if a too stringent preselection had been applied as early as phase 1 trials. Third, “surprise” responders can allow to learn more on the drug’s mechanism of action and target population than a well-selected homogeneous cohort of patients. Phase 1 trials are the only place where eligibility criteria can be flexible enough to enroll a variety of patients, and dedicated cohorts for rare tumor types or unselected patients should therefore be systematically added in the dose-expansion phase. Also, exceptional responders (or resistant patients) should be extensively sampled and studied, both in the tumor and peripheral blood—as well as lymph nodes if relevant—as such patients can be extremely informative. Of note, it is somehow worrying to observe how much the number of companies following the same targets has been increasing over the last 3 years (from 26 to 36%), while the number of targets pursued by only one company has been reduced from 42 to 26% [44]. Although no specific data is currently available for immune therapies in oncology, most immuno-oncology drug companies are currently following the same route and do not only develop agents directed against the same molecules but also target the same patient populations. Beyond making recruitment more difficult for phase 1 units, secondary to competing trials, this “herd mentality” also constantly reduces accessibility to novel drugs for the same patient populations (such as sarcoma, brain tumors, or rare diseases) and harms real and original innovation.
32.2.6 Response and Efficacy Assessment
Considering their mechanism of action, immune therapies can have unusual profiles of responses, including dissociated responses, delayed responses, hyperprogressions, and pseudoprogressions (initial increase in tumor size followed by tumor shrinkage) [45]. These correspond mostly to the fact that immune activation can take time, leading to a delay in onset of clinical effect, and that immune cells recruited to the tumor bed may cause an increase in tumor volume before it effectively shrinks [46]. Therefore, traditional World Health Organization (WHO) and Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1) criteria do not appropriately apprehend responses observed with immune therapies. Novel criteria, named immune-related response criteria (irRC), have therefore been developed [46] and added to the traditional RECIST criteria in most of the recent phase 1 trials evaluating immune checkpoint blockers. These better allow taking into account the different patterns of atypical responses observed with immune checkpoint blockers, including the potential for appearance of new nontarget lesions, growth of existing lesions preceding tumor shrinkage, and prolonged stable disease—sometimes ultimately followed by delayed response, even after treatment cessation. For example, 7.3% (24/327) of the melanoma patients treated with the anti-PD-L1 agent pembrolizumab in the Keynote-001 study presented atypical responses, including 4.6% (15/327) early pseudoprogressions and 2.8% (9/327) delayed pseudoprogressions [47]. Further, 14% (84/592) of the patients experienced progressive disease per RECIST v1.1 but nonprogressive disease per irRC. Also, the 2-year overall survival rates were 78% in patients with nonprogressive disease per both criteria, 37% in patients with progressive disease per RECIST v1.1 only, and 17% in patients with progressive disease per both criteria. This suggests that, at least in this population, using RECIST v1.1 may cause premature treatment interruption and lead to underestimation of the immune checkpoint blockers’ benefit. Although atypical patterns of response appear to be more frequent in melanoma patients and the benefit of using irRC criteria has been poorly validated outside this population, most phase 1 protocols evaluating immune therapies now recommend using (1) RECIST v1.1 criteria for response assessment and (2) irRC for treatment cessation. This means that confirmation of the progression is required by a second imaging assessment 4 weeks after the initial progression has been diagnosed, prior to taking a patient off-study. iRECIST criteria, which should become available soon, aim at merging irRC and RECIST v1.1 criteria in order to offer a widely implementable classification that better takes into account these atypical response profiles. Finally, provisions should be made to allow pursuing treatment beyond confirmed progression in case of clinical benefit (pending written approval from the study promotor, from the patient, and based on a consensus among all co-investigators). In line with these considerations, non-progression rate or clinical benefit rate might represent potential relevant alternatives to assess immune therapy efficacy in phase 1 (Fig. 32.4).
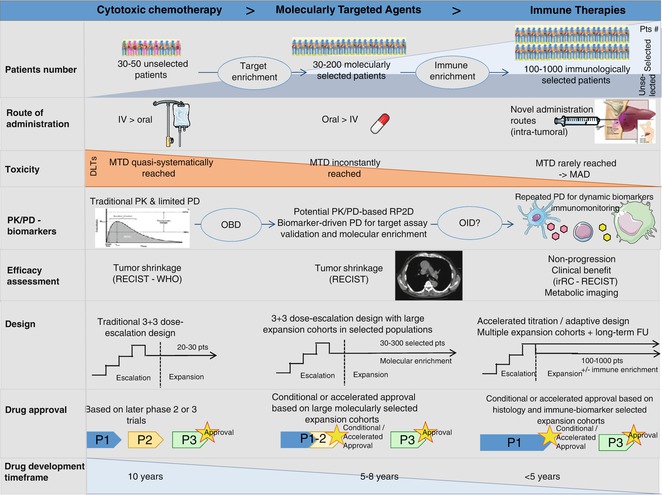
Fig. 32.4
The evolving landscape of phase 1 trials from cytotoxics to the immuno-oncology era (adapted from Postel-Vinay et al., with permission of the Editorial Office from Annals of Oncology). Striking changes have been observed in the landscape of phase 1 trials since the era of cytotoxic chemotherapy. Trials have moved from small-sized trials enrolling molecularly unselected patients to large-scale studies of several hundreds of molecularly—or phenotypically—selected patients enrolled in parallel expansion cohorts. Novel routes of administration have been developed as well as novel methods for dose recommendation, as the MTD is inconstantly reached. Pharmacodynamic and pharmacokinetic parameters play an increasingly important role, notably in helping determining the recommended dose for later phase trials. Whether, by analogy with the optimal biological dose of targeted agents, an optimal immunological dose based on PK and PD data of immunological monitoring could be used for immune stimulatory agents, which warrants further exploration. Early phase trial designs have evolved in parallel (also see Fig. 32.2), in order to maintain a rapid dose-escalation phase and assess efficacy in large selected cohorts. Consequently, the traditional phase 1–phase 2–phase 3 drug development strategy is being replaced by large phase 1/3 trials, which bypass the traditional phase 2 studies. All these evolutions have allowed dramatically reducing the drug development time, for patients’ benefit. #: number, IV intra-venous, MAD maximum administered dose, MTD maximum tolerated dose, OBD optimal biological dose, OID optimal immunological dose, P1 phase 1, P2 phase 2, P3 phase 3, PD pharmacodynamics, PK pharmacokinetics
Overall, it is likely that current criteria—either RECIST, irRC, or iRECIST—will be insufficient to recapitulate all peculiar patterns of response observed with immune therapies. Therefore, alternative strategies should be systematically envisioned and implemented in phase 1 trials. These include dynamic assessments (such as tumor growth rate), metabolic imaging, and also peripheral immunomonitoring—which could also be an indicator of response. Finally, whether overall survival should be systematically recorded in phase 1 trials of immune therapies is a current question. This endpoint has traditionally not been registered in early phase trials, as these primarily aim at assessing safety. Preliminary efficacy data, a secondary endpoint, was mostly assessed by the response rate or progression-free survival (PFS). Considering the effect of immune therapies on “raising the tail of the curve,” overall survival is a relevant endpoint to assess their efficacy. However, phase 1 trials, even within their current transformation to “phase 1 registration trials,” are not the place to assess overall survival, because: (1) phase 1 trials do not contain any control arm; (2) patients are likely to receive many lines of therapy thereafter, which will represent as many confusion factors; (3) this uselessly increases the workload associated with phase 1 trials, especially as phase 1 patients are often referred from external centers and go back to their hospital of origin after trial completion; and (4) as phase 1 studies arrive more and more early in the patient’s treatment lines, this data will very likely only be available once trial results have already been reported and published and therefore will virtually never be communicated.
Related posts:
 Peptide-Based Therapeutic Cancer Vaccines
Peptide-Based Therapeutic Cancer Vaccines
 The Human Tumor Microenvironment
The Human Tumor Microenvironment
 PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
 Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells
Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells
 Immune Checkpoint Inhibition in Lung Cancer
Immune Checkpoint Inhibition in Lung Cancer
 Immune Monitoring of Blood and Tumor Microenvironment
Immune Monitoring of Blood and Tumor Microenvironment
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


