Fig. 41.1
Schematic representation of analyses of patients’ blood and tissue specimens. Various methods are used to analyze blood, other body fluids, or solid specimens obtained by surgery or biopsy procedures
41.2 Flow Cytometry and Protein Expression Analysis
During the last two decades, characterization of immune cells has gained importance, providing key information in research and clinical studies. A variety of techniques is used to study the patient’s T cells (Fig. 41.2). The enormous technical progress during the last years in the field helped to build reliable instruments and to standardize technology and methods for analyzing cells and liquids from different origins, e.g., blood, other body fluids, and various tissues including tumors and lymphoid structures. The big variety and diversity of human samples, and the multiplicity of different cells, factors, cytokines, and chemokines, makes an imperative need for different techniques and dedicated procedures, in order to obtain maximum information from a limited amount of biological material.
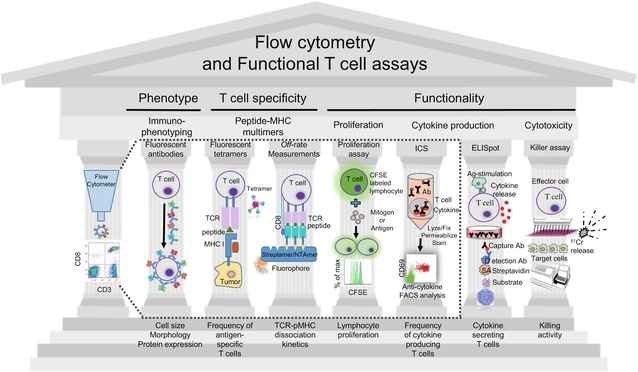
Fig. 41.2
Overview of the T cell assays. The illustrated techniques allow to profile and monitor T cell responses, providing information about phenotype, specificity, proliferation, and functionality of tumor-specific T cells. From the left to the right, flow cytometry enables rapid analysis of physical properties (e.g., size) and multiple molecular characteristics of individual cells, isolated from patient’s blood or the tumor microenvironment (TME). Antibodies conjugated to fluorescent dyes are used to label specific proteins on the cell membranes or inside cells. Antigen-specific T cells can be visualized using fluorescently labeled tetrameric peptide-MHC (pMHC) complexes that bind specific T cell receptors (TCRs). Besides determining the frequency of T cells, they can also be phenotypically characterized when combining tetramers with various antibodies. Novel approaches also allow the direct and precise quantification of TCR:pMHC dissociation rates (k off) on living CD8 T cells by real-time flow cytometry (using NTAmers) and/or microscopy (using Streptamers). Fluorescent CSFE labeling is used to assess T cell proliferation in vitro. Intracellular cytokine staining (ICS) is a very useful and widely used flow cytometry-based assay which detects the production and accumulation of intracellular cytokines after cell stimulation, as well as the frequency of the cytokine-producing T cells. Important non-cytometry-based assays are depicted to the right of the immune monitoring “Parthenon.” The IFN-γ ELISpot assay, a highly sensitive immunoassay, reveals the frequency of tumor-specific lymphocytes by forming one spot per cell upon antigen stimulation. Cytotoxicity and lytic capacity of lymphocytes is measured by the killer assay, whereby radioactive 51Cr is released from labeled target cells killed by cytotoxic T cells
The profiling and monitoring of immune responses are key elements in the development of new therapies against cancer and the evaluation of their efficacy. To monitor and quantify an immune response against tumors, it is important to use procedures and techniques (Fig. 41.2) that can reliably detect and characterize tumor antigen-specific T cells, provide information about their phenotype, their specificity, and their proliferation, as well as directly measure their various functions [1].
41.2.1 Flow Cytometry
Flow cytometry is one of the most broadly used technologies for the analysis of cells from blood and the tumor microenvironment (TME). This technology is used since about 30 years and is now widely adopted. Combinations of highly specific, fluorescent-labeled antibodies are used to identify various different cell types such as T, B, and NK cells, dendritic cells (DCs), and monocytes from a few microliters of blood [2]. The technique allows to enumerate the frequencies of the different cell types. Besides, one can determine many different qualities, for example, the cell’s activation and differentiation stages [3], or expression of various receptors by highly defined cell subsets [4]. It has been published that the two surface receptors CCR7 and CD45RA are differentially expressed in human T cells, depending on whether they are naïve cells (CD45RA+ CCR7+), memory cells (CD45RA− CCR7+), or effector cells (CD45RA−/+ CCR7−) [5]. Fluorescent labeling of additional markers such as the co-stimulatory receptors CD27 and CD28 allows to further classify T cells into distinct differentiation stages (Fig. 41.3). Modern flow cytometers have multiple (usually 12–18) fluorescent channels, allowing to simultaneously detect many further parameters such as the expression of activatory and inhibitory receptors, cytokines and chemokines, and (cytotoxic) proteases (Fig. 41.3).
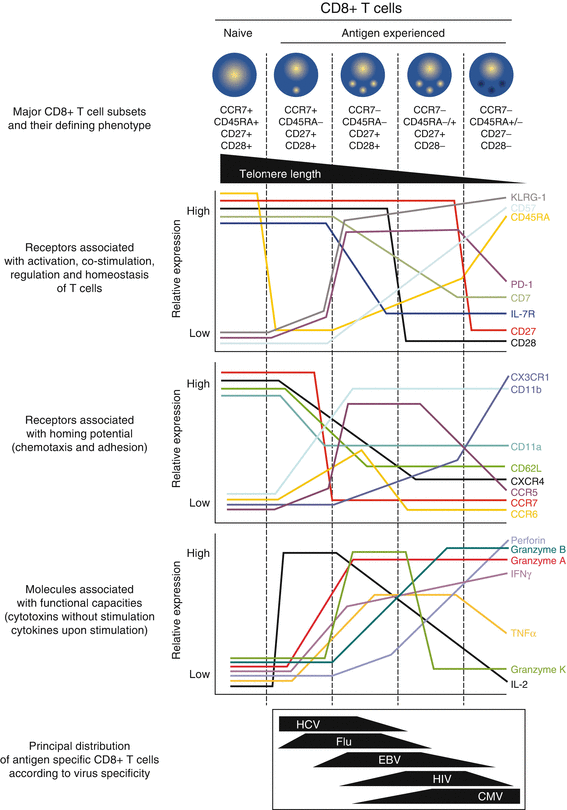
Fig. 41.3
Phenotypic associations within CD8 T cell subsets in humans and relationship with functional attributes. Five distinct subsets of circulating CD8 T cells are defined according to the expression of CD27, CD28, CCR7, and CD45RA. Relative telomere length and expression of a variety of cell surface receptors and intracellular molecules (related to T cell activation, costimulation, regulation, homeostasis, homing potential, and functional capacities) are illustrated in these subsets in a “resting” state according to data from the literature. The common phenotypic distribution of virus-specific CD8 T cells is also depicted, after clearance of the virus (Flu) or in latent infection stages (for HCV, EBV, HIV, and CMV). Flu influenza, HCV hepatitis C virus, EBV Epstein-Barr virus, HIV human immunodeficiency virus, CMV cytomegalovirus. The figure is taken from the paper by V. Appay et al. [3], Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A, 2008 vol 73 (11):975
Most practical is the analysis of peripheral blood mononuclear cells (PBMC) which are obtained by gradient centrifugation of freshly obtained venous blood. This technique eliminates red blood cells and granulocytes, giving the advantage to analyze a purified cell population that can also be frozen in aliquots and thawed later on for further investigation. But researchers become more and more aware of the fact that this strategy not only leads to the loss of potentially important cell types but also introduces small artifacts, meaning that some cell populations change their characteristics and thus give rise to misleading results. Therefore, many cell types should rather be analyzed using fresh whole blood, as, for example, shown for myeloid-derived suppressor cells (MDSCs) [6].
Huge efforts have been made in developing new technologies to increase the information, particularly when patients’ specimens are small and/or rare. One strategy is flow cytometry with mass spectrometry where antibodies are labeled with heavy metal ion tags, rather than fluorochromes. Here the readout is done by time-of-flight mass spectrometry (Cytof/Fluidigm) [7]. Another strategy is to combine flow cytometry with microscopy. The Image Stream machine (Amnis) is one example of an instrument where fluorescent-labeled cells can be analyzed and identified by their fluorescent molecules like with flow cytometry at the cell surface, and at the same time, a camera is taking pictures of the cells. This allows determining whether the distribution of the fluorescent-labeled molecules is homogeneously distributed over the surface of the cells or polarized in distinct areas or compartments of the cells. The fact that the cells pass with a lower speed through the instrument allows even to observe when cells are in contact to each other and to identify eventual exchange of molecules in their contact zones [8].
Furthermore, new live imaging systems like, e.g., IncuCyte (Essen BioScience), CytoSMART (Lonza), and Operetta (Perkin Elmer), allow to observe cells and their activity in vitro under “online” in culture conditions. In these technologies cameras are placed in incubators and take photos at defined time points over several days under optimal conditions. This allows, for example, observing movements of cells in presence of target cells, e.g., tumor cells or chemotaxis movements of cells due to external stimulation [9]. One of the latest technical developments is the Chip-Cytometer (ZellkraftWerk). This technique allows fluorescent microscope analysis on cells in suspension and on tissues fixed on microscope devices which can be re-stained multiple times with different markers on different cells or on the same cells [10]. The technique still requires optimization but could eventually replace cell consuming flow cytometry with time-consuming immunohistochemistry (IHC). The advantage of this technology is the analysis of rare material/cells for different cell surface and intracellular molecules with the strength of fluorescence detection well known from fluorescent analysis by flow cytometry.
41.2.2 Protein Arrays
Another possibility to obtain information about the differential states of cells and to compare samples from healthy donors with those from patients at different time points is to perform protein arrays, which can be done of bulk material or of distinct cell types previously sorted by flow cytometry or magnetic beads. This technology allows qualitative protein profiling and can also characterize protein phosphorylation revealing intracellular signaling activities. The technique is promising for screening for key factors and eventually identifies candidate biomarkers [11].
41.2.3 Cytokine Analysis
T cells have the capacity to secrete one or even multiple cytokines. The detection of soluble molecules secreted by different cell types from in vitro experiments or directly from the serum of blood samples is possible by standard ELISA assays. This technique is simple and efficient but has a relatively low sensitivity and can only analyze one specific molecule at the time. New sophisticated technologies have been developed for very sensitive detection of multiple cytokines, chemokines, or other molecules, based on either bead capture assays with liquid-based systems (Flow cytometry, Luminex) or on imaging systems, where the secreted molecules are captured on specifically coated membranes, e.g., MSD (Meso Scale Discovery). These techniques also have the advantage that they can detect multiple molecules simultaneously from a small amount of sample [12].
41.2.4 Bioinformatics Support for Data Analysis
Conclusive analysis of huge datasets coming from one or even multiple analytical approaches is challenging. Analyzing data from flow cytometry is tainted even when respecting commonly established and firm rules. For example, subjective gating decisions can strongly influence the results and their interpretation. A more objective way to analyze such data is by using novel dedicated software programs. Mathematic algorithms are capable to analyze and compare datasets in an unbiased way. By combining all different parameters, deep analysis of datasets is possible. Typically, cell populations can be rapidly identified, and even unconventional cells are found that might have been ignored with conventional cell gating.
41.3 Further Assays to Monitor T Cell Functions
Besides flow cytometry, there are various other techniques to analyze T cell functions. Several standard functional T cell assays exist for determining cytotoxicity [13], cytokine production, and proliferation [14], as overviewed in Fig. 41.2. Moreover, assays that can simultaneously detect T cell frequency and function, such as the interferon-γ (IFN-γ) enzyme-linked immunospot assay (ELISpot), have gained increasing popularity for monitoring T cell responses in clinical trials and in basic research.
41.3.1 Analyzing Cell-Mediated Cytotoxicity
T cell-mediated cytotoxicity represents a key mechanism in the immune response to tumors. Therefore, monitoring methods for the appropriate assessment of cytotoxic immune reactivity is thought to be crucial [15]. A classical and popular assay for evaluating cell-mediated cytotoxicity by T lymphocytes (CTLs) and by natural killer cells is the [51]Chromium (51Cr) release assay or cytotoxicity assay, schematically depicted in Fig. 41.2. First developed in 1968 by Brunner et al. [16], it continues to be widely used for testing killing activity in immune monitoring laboratories. It is based on the passive internalization and binding of the soluble radioactive chromium from sodium chromate by target cells in single-cell suspensions. Subsequent lysis of the target cells by added effector lymphocytes results in 51Cr release into the cell culture supernatant, which is then detected by a gamma counter. Although several modified alternatives have been introduced [17], this assay remains a “gold standard” to measure cell-mediated cytotoxicity [18].
However, there are several disadvantages that create a need for more accurate methods: this assay has a relatively low level of sensitivity, often one needs to stimulate cytotoxic lymphocytes several times before they reach detectable levels of lytic activity, which unfortunately alters the composition and activity of the original T cell populations, and it does not provide direct information about the in vivo function and the behavior of single effector cells. Furthermore, there is evidence that the killing activity may largely differ between individual T cells and that “super killers” may dominate in cytotoxicity assays [19], meaning that many other T cells do not contribute significantly to target cell killing. Some target cells label poorly with 51Cr, and other target cells show high spontaneous 51Cr release. Since autologous tumor cells are difficult to obtain, surrogate targets must be used, but they may not reveal the actual ability of lymphocytes to lyse autologous tumor cells in vivo. Finally, the inter-assay variability is considerable, and there are biohazard and disposal problems associated with radioisotope usage.
41.3.2 The ELISpot Technique for Assaying Cytokine- and Interferon-Producing Cells
The IFN-γ ELISpot assay is a highly sensitive immunoassay that reveals tumor-specific lymphocytes by forming one spot per cell provided that it secretes IFN-γ in response to antigen stimulation [15]. This assay is well suited to determine the frequency of functional cells. Versteegen et al. [20] were the first to use the ELISpot to detect human cells secreting IFN-γ. Subsequently, an important modification of the ELISpot assay was made by employing nitrocellulose membranes and specific monoclonal antibodies, increasing reproducibility and sensitivity of the assay [21]. With the recommendation of the 13th International AIDS Congress in the year 2000 to use the ELISpot technique due to its performance for immune monitoring purposes, it became one of the most important direct ex vivo methods in cellular immunology [22, 23].
Clinical trials and immune monitoring require comparability and reproducibility of results. The ELISpot assay is well suited, because it is a relatively simple procedure and accurate for testing any T cell, including those directly withdrawn from patients without any in vitro amplification or modification, thus best reflecting their in vivo state. Therefore, the ELISpot assay has become a standardized and validated method. The first steps to harmonize the process for widespread applications included standardization of protocols, materials, and reagents [24], for instance, by using pre-coated 96-well plates [25]. Consequently, the inter-assay and intra-assay variability was minimized, and precision and reproducibility were increased.
Schematic illustration of the principle of the ELISpot assay is summarized in Fig. 41.2. Each spot corresponds to an individual cytokine-secreting cell. The spots are counted by an ELISpot Bioreader, allowing to calculate the frequency of functional T cells. The ELISpot assay captures the presence of cytokines of interest, immediately after secretion from the “spotting” cells, in contrast to measurements that are skewed by receptor binding or protease degradation. The assay is considered as one of the most sensitive T cellular function assays available. The limit of detection typically achieved can be 1 in 100,000 cells with standard ELISpot assays and with an average of less than 1 in 200,000 PBMC in modified assays [26]. The assay is particularly useful when only small cell numbers are available.
Classically, this assay is performed with a single (high) peptide dose, usually at the concentration of 1 micromolar. Alternatively, or in addition, one can use titrated amounts of peptide to generate a characteristic sigmoid dose response curve (Fig. 41.4), used to determine the functional avidity of T cells. The mean “functional avidity” corresponds to the peptide concentration that confers 50% maximal activity (EC50), i.e., the amount of antigenic peptide required for half-maximal IFN-γ ELIspots. Because antigen is often rate limiting in vivo, the functional avidity is a parameter that is particularly important for evaluating the functional potency of antitumor T cells. The functional avidity depends on TCR-peptide-MHC interactions on the T cell surface, where the TCR clustering, the involvement of the CD8 co-receptor, the local concentration of adhesion molecules, and the involvement of co-activating and co-inhibitory receptors are all important parameters that modulate the strength of the intercellular interaction and thus T cell activity [27–29]. The functional avidity refers to the accumulated “dynamic strength” of multiple affinities of individual non-covalent binding interactions [30]. An additional parameter is the functional avidity heterogeneity, revealed by the slope of the curve (Ioannidou K, Baumgaertner P, Gannon PO, Speiser MF, Allard M, Hebeisen M, Rufer N, Speiser DE. Heterogeneity assessment of functional T cell avidity. Scientific Reports. 2017 Mar 13;7:44320. doi: 10.1038/srep44320).
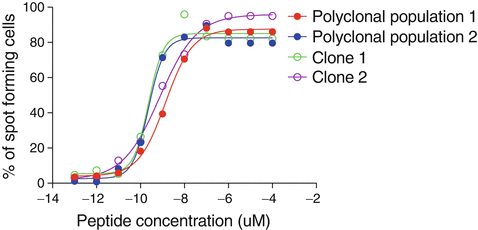
Fig. 41.4
Examples of ELISpot titration curves from two individual CD8 T cell clones and polyclonal T cell populations from two patients. Each microculture was prepared with 300 antigen (Melan-A)-specific T cell clones or polyclonal T cell populations that contained 300 antigen-specific cells. The percentages of antigen-specific T cells in the polyclonal populations were previously determined with fluorescent tetramers by flow cytometry, allowing to calculate how many cells had to be plated to have 300 antigen-specific T cells per microculture. For stimulation, all microcultures were supplemented with 20,000 T2 cells and the indicated concentration of antigenic (Melan-A) peptide. Ten concentrations of peptide were tested, each in triplicates, thus requiring 30 microcultures (in 96-well plates). Spots per well were counted with the ELISpot Bioreader 5000, and curves were plotted using Prism software
41.3.3 Intracellular Cytokine Staining
Intracellular cytokine staining (ICS) revealed by flow cytometry is one of the most popular assays in the immunologists’ toolbox, designed to assess complex T cell responses. One of the specific advantages of ICS is that it enables the simultaneous assessment of multiple phenotypic, differentiation, and functional parameters, including the expression of multiple cytokines [31]. ICS is used in combination with other flow cytometry protocols for immunophenotyping using fluorescent antibodies specific for cell surface markers and/or with fluorescent MHC multimers to detect antigen-specific T cells, making it an extremely flexible and versatile method. The main experimental steps of ICS are that the cells of interest are activated using either a specific peptide or non-antigen-specific activation by, e.g., anti-CD3 antibody or a mitogen. In the next step, an inhibitor of protein transport (e.g. brefeldin A) is added to retain the cytokines within the cells. After washing, the antibodies specific for cellular markers are added. The cells are then fixed (e.g., with paraformaldehyde) and permeabilized, and the anti-cytokine antibodies are added. Finally, the cells are analyzed by flow cytometry (Fig. 41.2). Many technical advances have been achieved, e.g., the polychromatic assays, designed to detect five or more separate functions of T lymphocytes (e.g., the production of cytokines and chemokines and the detection of degranulation as surrogate for cytotoxicity) while simultaneously identifying multiple surface markers [32].
41.4 Multiplexed Immunohistochemistry (IHC) Assays
Histology and IHC are excellent methods to approach the great complexity of biological processes in various tissues. For cancer patients, major challenges are to understand the disease driving biological mechanisms in the TME. Microscope-based tissue analysis allows the identification of structural and functional molecules and their intra- or extracellular localization. Analyses can be done with freshly frozen (FF) or formalin-fixed paraffin-embedded (FFPE) tissue samples. Their appropriate and timely handling is critical [33, 34] in order to avoid unnecessary protein and nucleic acid damage. Careful measures must be taken in order to optimally handle tissues such that the integrity of biomolecules is maintained, and detailed knowledge is obtained about eventual procedural artifacts. Also, reference tissues are required for quality control, including the verification whether the staining is capable to reveal the targeted markers.
IHC techniques use antibodies for specific labeling and staining of molecules in tissue sections. Antibody binding can be detected with an enzymatic reaction that induces chromogenic precipitation (to stain for maximally three to four markers on a single tissue section), or by fluorescent dyes (up to about eight markers). Dedicated signal amplification techniques are used to covalently link antibodies with dyes, minimizing cross-reactions upon the required subsequent rounds of staining with additional antibodies [35]. The markers can be identified with a multispectral camera. Considerably, larger numbers of markers (possibly >100) can be applied with novel techniques based on successive cycles of staining, scanning, and then removal (or bleaching) of the fluorophore, which however also requires specific software that can subsequently overlay the serial images [36, 37].
IHC is usually done on slices carrying conventional tissue sections. An elegant alternative technique is the so-called tissue microarray, which consists of punching out cylinders (of 0.6–2 mm diameter) of tissue paraffin blocks of microscopic regions of special/specific interest, customized according to hypothesis and experimental design. A large number of tissue cylinders are then used to produce a new paraffin block, in which these tissues from up to >100 patients are placed. Slices of such tissue microarray blocks enable to stain and analyze large numbers of tissues much more efficiently, favoring large-scale quantitative investigations [38, 39].
Traditionally, analysis and interpretation of histological images are evaluated qualitatively and semiquantitatively with the microscope by the eyes of a trained pathologist. Novel automated image analysis software is increasingly used to automate and accelerate this process, enabling the evaluation of large tissue regions in a fully quantitative manner [40]. However, this still requires the supervision by pathologists, as the interpretation of tissue complexity must always be quality controlled.
Overall, the use of efficient and precise methods for the investigation of tissues represents a central pillar for both research and patient assessment. For example, this is needed for the quantification of tumor-infiltrating T lymphocytes (TILs), which are often of high prognostic and predictive value and can even be more important than the traditional TNM (tumor, node, metastasis) classification of tumors. These techniques allow quantifications in defined areas, e.g., in tumor cell nests, invasive margins, or peri-tumoral regions, each of which may have different causes and consequences [41, 42].
41.5 High-Throughput Techniques
Several techniques can determine thousands of parameters and are therefore called high-throughput techniques (Fig. 41.5), particularly the approaches that are sequencing based. Whole exome sequencing (WES) can be used to identify gene abnormalities in tumor cells, revealing specific mutations and overall mutational load. A comprehensive overview of gene alterations in tumor cells is obtained when WES is combined with DNA copy number profiling [43], revealing gain and loss of function alterations. In addition, an increasingly important technique is RNA sequencing (RNA-seq), enabling quantitative assessment of the expression of mRNA or microRNA. RNA-seq can be used for very many different purposes, for example, for gene signature studies, or the characterization of chemokines, cytokines, and other immune factors [43–48]. Furthermore, DNA methylation profiling is used to study epigenetic mechanisms that regulate immune gene expression, or cell lineage-specific epigenetic modifications, also permitting estimations of the percentages of the different cell populations present in the microenvironment [49–51].
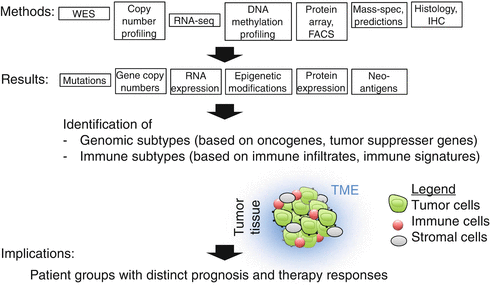
Fig. 41.5
 Peptide-Based Therapeutic Cancer Vaccines
Peptide-Based Therapeutic Cancer Vaccines
 The Human Tumor Microenvironment
The Human Tumor Microenvironment
 PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
 Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells
Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells
 Immune Checkpoint Inhibition in Lung Cancer
Immune Checkpoint Inhibition in Lung Cancer
 Immunogenic Stress and Death of Cancer Cells in Natural and Therapy-Induced Immunosurveillance
Immunogenic Stress and Death of Cancer Cells in Natural and Therapy-Induced Immunosurveillance




High-throughput approaches to characterize cancer cells and their microenvironments for improving patient diagnosis and treatment, toward precision medicine. An increasing number of techniques allow comprehensive characterization of the various tissues and cells from cancer patients, enabling detailed characterization of tumor biology and immune parameters in primary and metastatic cancers. TME tumor microenvironment, WES whole exome sequencing, RNA-seq RNA-sequencing, IHC immunohistochemistry
Related posts:
Peptide-Based Therapeutic Cancer Vaccines
The Human Tumor Microenvironment
PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells
Immune Checkpoint Inhibition in Lung Cancer
Immunogenic Stress and Death of Cancer Cells in Natural and Therapy-Induced Immunosurveillance
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
