Fig. 13.1
A stepwise development of CRC includes ACF, adenoma, and adenocarcinoma formation which eventually metastasize. ACFs preneoplastic lesions are develop when an insult occurs in the colonic mucosa upon carcinogen treatment in rodents, and few of these develop into adenomas and adenocarcinomas. During this process, the NK cells activity towards tumor cell is inhibited by increased number and activity of Tregs with increased activity of COX-2 and 5-LOX
13.4.4 Immune Responses During PDAC
In pancreatic ductal adenocarcinoma (PDAC), a decreased number of NK and NKT cells are observed in patients with more aggressive tumor growth, and the numbers of these cells are inversely correlated with disease progression. Clinical samples of PDAC showed no NK cell infiltration, but significantly high numbers of macrophages were observed in patients with metastatic disease spreading to lymph nodes (Fig. 13.2). The presence of inflammatory cells contributes to the angiogenic phenotype in PDAC. Preclinical in vivo and in vitro studies have shown that IFN-α has a direct toxic effect on PC cells. A phase III trial combining chemoradiotherapy with IFN-α showed immediate activation of antigen-presenting cells (APCs) and NK cells followed by antigen-specific activation [30]. It is evident from these reports that restoration of NK cells and their functions by administration of immunomodulatory agents leads to enhanced cellular immune response against PC in clinical and preclinical models. PC cells can produce cytokines (including transforming growth factor-β (TGF-β), IL-10, and IL-6) and express surface antigens, such as programmed death-1 (PD1), indoleamine 2,3-dioxygenase (IDO), vascular endothelial growth factor (VEGF), and Fas-L that suppress immune responses against PC. Immune cell infiltrates consisting of tolerogenic DCs, tumor-associated macrophages (TAMs), and Tregs facilitate tumor growth and metastasis [31, 32] (Fig. 13.2).
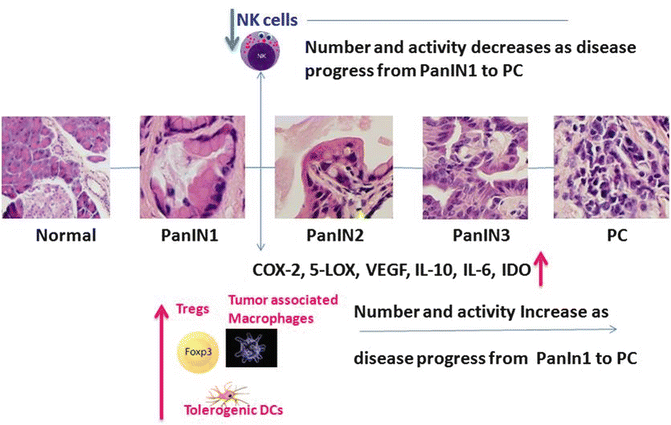
Fig. 13.2
Immune cell modulation during PanINs progression to PC. During stepwise PanIN 1 progression to PanIN 2, Treg number and activity increase, along with other immune cells such as tumor-associated macrophages, which makes Dcs tolerogenic towards PanIN 3 into PC progression. During this tumor initiation process, NK cell activity and number decrease which eventually helps the lesions to progress to PC. Also arachidonic acid metabolites, angiogenic factors, and cytokines (COX-2, VEGF, IL-10, IL-6 and IDO) are highly expressed and cooperate during PanINs progression to PC
CD8+ T cells are the predominant immune cells found in pancreatic cancer. However, downregulation of activation markers on CD8+ T cells is reported to reduce their cytotoxic effects towards tumor cells, suggesting that these cells become ineffective against tumors. Tregs, IL-6, and transforming growth factor (TGF)-β1 levels are high in locally advanced metastatic pancreatic cancer patients, and an increased number of Tregs was observed in peripheral blood of patients who had progressive pancreatic tumor growth. Hence, in PDAC, a high prevalence of Tregs seems to be a marker of poor prognosis [33]. These cells also were observed among tumor-infiltrating cells during multistage development of PDAC. In general, naturally occurring (n)Tregs help in developing self-antigen tolerance by suppressing NK cells, NKT cell activation, and maturation of DCs. DCs help to balance tolerance to self with elicitation of immunity towards tumors. DCs are reported to activate pancreatic stellate cells by increasing their chemokine production [34, 35], migration, and expression of toll-like receptors (TLRs). Infiltrating DCs in chronic pancreatitis exacerbate inflammation, which can lead to PC. Induced Tregs (iTregs) are responsible for regulating immunity against foreign (microbial) and tissue antigens. Tumor cells induce cytokines (IL-2, IL-10, TGF-β) and other enzymes (cyclooxygenase (COX)-2, 5-lipoxygenase (LOX), IDO) that can enhance production of Tregs and, thus, help the tumor escape from immune surveillance (Fig. 13.2). The observation of depleted Tregs helps to provide a mechanism for preexisting immune suppression by them against the tumor despite the presence of tumor antigens. The evidence from both preclinical and clinical studies supporting a critical role of Tregs in immune suppression and evasion by tumors suggests the importance of targeting Tregs or modulating their functions with preventive and therapeutic agents.
13.4.5 Immunomodulatory Effects of NSAIDs
Multiple endogenous mediators regulate differentiation, maturation, and activation of various immune cells. These include various cytokines and chemokines, bioamines, purines, and COX-/LOX-mediated production of lipid mediators derived from arachidonic acid (AA) such as prostaglandins (PGs) and leukotrienes (LTs) that are recognized to have immune regulatory effects. NSAIDs, including salicylates (aspirin), propionic acid derivatives (ibuprofen, naproxen), acetic acid derivatives (sulindac), enolic acid derivatives (piroxicam), and selective COX-2 inhibitors (celecoxib), inhibit COX-1 and/or COX-2 and LOX pathways. Thus, they inhibit the production of PGs that contribute to pain and inflammation and are widely used for treating arthritis. They function as anti-inflammatory agents, but they may exert immune modulatory effects differentially on macrophages and lymphocytes.
COX-1 and COX-2 act on unsaturated 20 carbon essential fatty acids such as AA and generate prostaglandin H2 (PGH2). Cell-specific PG synthases catalyze the conversion of PGH2 to five primary bioactive prostanoids PGD2, PGE2, PGF2α, PGI2, and thromboxane A2 (TXA2). Each of these prostanoids is generated in macrophages and DCs. The prostanoids that are downstream of the COX-2 pathway influence many functions of cells involved in the immune system such as macrophages, DCs, NKs, and T and B cells and play an important role in the physiology and pathology of immunologic responses during inflammation, tumor growth, and development [36, 37]. The role of PGE2 in these responses is prominent; it reduces activity of NK cells and CTLs by decreasing IL-2, IL-15, and, most likely, IL-2 production and expression of the IL-2 receptor on effector cells [36, 38, 39] (Fig. 13.3). It also reduces expression of IFN-γ by NK cells, thereby abrogating the NK cell helper function in mediating DC-induced Th1 responses [40]. Reduced NK cell function in response to PGE2 increases establishment of metastases in preclinical animals [41]. PGE2 also promotes the production of pro-angiogenic and immunosuppressive VEGF and monocyte chemotactic protein 1 (MCP-1) by mast cells [37, 42] and leads to an increase in Tregs (Fig. 13.3).
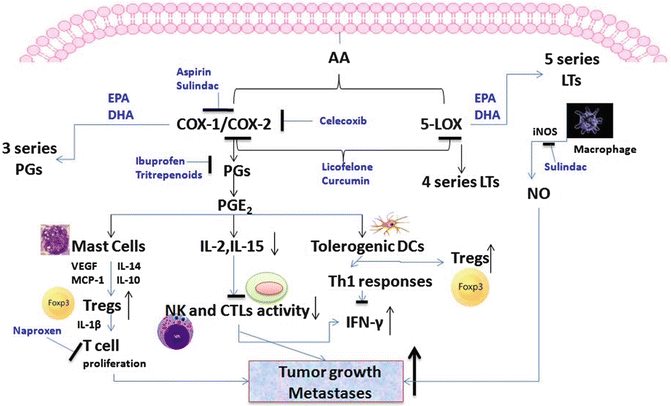
Fig. 13.3
Arachidonic acid (AA) metabolites are very well known for their tumor-promoting effects in colon and pancreatic cancer. Notably COX downstream molecule PGE-2 is reported to enhance tumor growth by helping in the development of tolerogenic DCs and Tregs and decrease NK cell activity. 5-LOX metabolites involve 4 series leukotrienes (LTs) which play vital role during tumor growth and progression. Formation of NO favors inflammatory conditions and helps in tumor growth and development. NSAIDs, COX-LOX inhibitors, and natural agents (triterpenoids, curcumin) are reported to inhibit COX-LOX activities and PGE-2 levels and inhibit tumor formation. Also, omega-3 fatty acids, EPA and DHA, modulate the COX-LOX activities by forming less potent metabolites such as three series PGEs and five series LTs
Roles of COX and LOX are well established in CRC, wherein generated PGs and other mediators may initiate and promote cancer by suppressing immune responses, triggering cell proliferation, inhibiting apoptosis, and stimulating angiogenesis. These enzymes are also responsible for increased production of the Th2 cytokines IL-14 and IL-10, which help to increase Treg number and activity (Fig. 13.3). In tumor-bearing mice, Tregs can help switch Th functions and contribute to the increased inflammatory conditions promoting cancer in polyposis conditions. 5-LOX metabolites such as LTB4 increase the activity of immune suppressor cells and increase growth of tumors.
Aspirin has received a lot of appreciation in preventive and therapeutic uses for its traditional anti-inflammatory properties and for having lifesaving activity against cardiovascular events such as heart attacks. In a retrospective cohort study, aspirin use was associated with reduced cancer risk in a subgroup of patients whose colon tumors were expressing COX-2. However, aspirin inhibits both COX-1 and COX-2 (Fig. 13.3) and can lead to gastrointestinal (GI) ulceration, bleeding, and hemorrhagic stroke. The GI risk lies between 0.1 % and 1 %. Modified forms of aspirin, including nitric oxide derivatives such as NO aspirin, are gaining much attention due to their gastro-protective effects. Inhibition of COX pathways may cause an increase in LOX catalytic pathways, and LOX metabolites may cause deleterious effects and may stimulate cell proliferation in tumors. It has been established that low doses of aspirin trigger synthesis of factors that promote resolution of inflammation, called aspirin-triggered lipoxins (APLs), such as 15R-epilipoxin A4. Several large epidemiological studies have shown that long-term, frequent aspirin intake is associated with a 50–60 % reduction in the risk of colorectal adenomas and carcinomas [43–45]. The authors’ preclinical results are consistent with inhibition of COX-2-mediated PGE2 synthesis by aspirin; NO aspirin resulted in inhibition of CRC in both mice and rats [46].
Aspirin may also have some additional immunopharmacological properties, although available information is limited. An in vitro study demonstrated no effect of aspirin on spontaneous or INF-β-stimulated NK cell activity [47]. Therapeutic doses of aspirin have been shown to increase selectively nTreg number and function in mice [48, 49]. Various preclinical evidence has shown that aspirin may have profound effects on maturation and differentiation of different DC phenotypes, causing DC tolerance and impairing antigen presentation by these cells. Aspirin-induced tolerogenic DCs may help in de novo generation of Tregs or in induction of regulatory functions in naïve T cells. These reports suggest that aspirin may cause immune suppression by directly enhancing Treg activity or indirectly by generating tolerogenic DCs. Hence, aspirin has potential to suppress immune responses in certain diseases such as autoimmunity. The authors and others have demonstrated the existence of Tregs in CRC during initial stages of the disease progression. Thus, although, meta-analyses and systematic reviews suggest that aspirin use is associated with a decreased incidence of colonic adenomas, colorectal cancer, metastatic colorectal cancer, and death due to colorectal cancer [50, 51], one needs to be cautious with the use of aspirin for cancer treatment in cases where immunosuppression by Tregs is observed. The potential benefit of aspirin in cancer is dependent on dose, timing, and number of years of use [44]. At least 7–10 years of aspirin use at 325 mg daily has been shown to decrease cancer. Similar estimates were made in a later meta-analysis of four placebo-controlled trials of aspirin; the beneficial effects of aspirin in reducing cancer risks were very consistent in various clinical trials [52]. Several intervention trials have evaluated the benefits of aspirin in prevention trials, and aspirin showed modest benefit on reducing recurrence of colorectal adenomas. These studies were conducted in patients already at the adenoma or cancer stages and not simply in patients having a risk of developing adenomas. Two meta-analyses have been published which suggest that the risk of recurrent adenomas is reduced by about 13–18 % [51, 52]. Trials focusing on prevention of colon cancer by preventing adenoma recurrence did not show consistent results with the use of aspirin [43, 53–59]. A recent report on regular use of aspirin in a cancer screening trial showed a reduced risk of hyperplastic and adenomatous polyps, with a slightly greater risk reduction in an age group of 70–74 compared with the 55–69 age group [60]. Overall, these trials suggest that prolonged use of aspirin reduces the risk of colon cancer. A recent study in 904 pancreatic cancer patients and 1,223 similarly matched healthy controls suggested that regular use of aspirin at least once a month resulted in 29 % reduced risk of developing PC. Those who took low-dose aspirin regularly had a greater 35 % risk reduction for PC. In a preclinical study, LsL-KrasG12D;LsL-Trp53R172H;Pdx1-Cre transgenic mice were randomly assigned to receive mock treatment, gemcitabine, or a combination of gemcitabine and aspirin; results showed suppression of Tregs in the mice receiving the combination treatment [61], which, in part, may have affected the increase in survival of diseased animals.
Ibuprofen shares with other NSAIDs such as aspirin the ability to inhibit the activity of COX-1 and COX-2, thus reducing the generation of pro-inflammatory stimuli. Lower ibuprofen drug concentrations act as activators of splenocyte proliferation, possibly through inhibition of thromboxane (TX)/hydroperoxyeicosatetraenoic acid (HETE) synthesis. Higher drug concentrations also activate splenocyte proliferation through inhibition of PGE2 and PGI2 [62]. Thus, both high doses and low doses of ibuprofen are able to regulate PG synthesis in cells (Fig. 13.3). Dietary administration of ibuprofen at 400 ppm resulted in a 41 % reduction of azoxymethane (AOM)-induced colon tumor incidence in F344 rats [63]. Ibuprofen caused a significant growth inhibition of HT29 xenografts overexpressing Rac1b (as occurs in serrated tumors having BRAF mutations), but the growth inhibition was independent of COX inhibition [64]. These findings indicate that the beneficial effect of NSAIDs in CRC may not rely solely on an anti-inflammatory response. Cameron et al. [65] reported that ibuprofen transformed noncytotoxic macrophages into cytotoxic ones and also enhanced the cytotoxic activity of macrophages in colon cancer patients [65]. A case-controlled study showed that, as with low-dose aspirin, intake of one or more pills per week of regular ibuprofen (200 mg) was associated with a significant (~68 %) reduction in the risk of colon cancer [66, 67]. A study in a large general risk population supports previous work showing that recent use of ibuprofen is associated with a decreased risk of colorectal adenomas [60]. Ibuprofen also shares with aspirin similar GI toxicity, which restricts the use of this drug to patients suffering from ulcerative colitis. Hence, a derivative of ibuprofen, phospho-ibuprofen, was designed and tested by the authors and others in AOM-induced colon cancer in F344 rats and was observed to have better efficacy in inhibiting development of aberrant crypt foci (ACF) and colon cancer compared with ibuprofen. The authors also tested phospho-ibuprofen in a transgenic mouse model of PC and found that it significantly inhibited progression from pancreatic intraepithelial neoplasias (PanINs) to PDAC.
NSAIDs have been shown to reduce spleen lymphocyte proliferation and to inhibit T-cell proliferation [68–70]. The S enantiomer of naproxen was shown to cause apoptosis of polymorphonuclear neutrophils (PMNs), which is a crucial mechanism for PMN, removal while resolving inflammation [71]. Immunotoxicology studies to assess the effects of naproxen on cell-mediated immunity showed no effect on NK cell activity [72]. Although high (supra-pharmacologic) concentrations of these drugs have been shown to induce some in vitro immunomodulatory effects on the innate immune system, they failed to show any effects in vivo. More detailed studies of naproxen effects in vivo are therefore needed to understand how it influences immunity in cancer patients.
Anti-inflammatory NSAIDs releasing NO (NO-NSAIDs) are a new class of anti-inflammatory drugs to which an NO-releasing moiety is added. NO-naproxen was developed with the goal of reducing the gastrointestinal toxicity associated with regular use of NSAIDs. These compounds have been shown to retain the anti-inflammatory, analgesic, and antipyretic activity of the parent compound but to be devoid of gastrointestinal (GI) toxicity [73]. Naproxen was reported to reduce IL-1β, but NO-naproxen reduced both IL-1β and TNF-α plasma levels [74]. NO-naproxen and naproxen both reduced the proliferation of T cells, whereas naproxen needed double the dose of NO-naproxen to show a similar function (Fig. 13.3). NO-naproxen has a greater effect on T-cell responses due to the presence of the NO moiety [74]. Both agents showed potent anti-inflammatory and antitumor properties in preclinical animal models of CRC.
ApcMin/+ mice genetically predisposed to spontaneous development of colon tumors who were fed low-dose naproxen for 45 days had 70.3 % fewer small tumors than the control animals, and mice fed low-dose NO-naproxen had 64.0 % fewer tumors than control animals. An 89.3 % reduction in microadenomas was observed when mice were fed high-dose naproxen. Relevant human serum concentrations of naproxen (1 μg/mL) were found to have chemopreventive effects in human colon polyp cells and to reduce the expression of signaling pathway genes (Ras, Fos, Myc, Jun, Vav1, ELK1, ELK4, multiple map kinases, and TGFB receptor (R) 1) implicated in cancer and growth [75]. Sulindac and naproxen, individually and in combination with atorvastatin, caused significant reduction in AOM-induced colon tumors in F344 rats and inhibited key inflammatory markers such as inducible nitric oxide synthase (iNOS), COX-2, and phospho-p65 as well as the inflammatory cytokines TNF-α, IL-1β, and IL-4. These studies also provided evidence for a strategy of NSAID combination with statins for reduced GI toxicity.
Sulindac has long been used to help prevent the development of intestinal polyps that can lead to colon cancer and is well established as a chemopreventive agent for CRC. Min mice receiving sulindac had fewer intestinal tumors [76]. In a randomized, placebo-controlled, double-blind study, administration of sulindac at dose of 300 mg/day for 6–12 months resulted in complete eradication of polyps in patients with familial adenomatous polyposis (FAP) [77]. Sulindac and several other NSAIDs were able to control growth of desmoid tumors in patients affected by FAP or Gardner’s syndrome [45, 78–80]. A significant reduction in the size of adenomas was reported in FAP patients after long-term therapy with sulindac [77, 79–81]. As in animal studies, sulindac was more effective when given at early stages of tumor development. Preclinical in vivo studies showed that sulindac inhibits dimethylhydrazine (DMH)-induced colon tumor incidence and multiplicity in mice when administered in the diet throughout the period of carcinogen administration, but not when given 17 weeks after DMH administration [82, 83]. In another study, sulindac (10 mg/kg body weight administered twice daily by gavage) inhibited DMH-induced primary colon tumor development and growth in rats. Ahnen et al. [84] showed that dietary administration of sulindac and its metabolite sulindac sulfone significantly inhibited AOM-induced colon carcinogenesis in F344 rats [84]. Sulindac inhibits COX activities as well as polyamine synthesis, for which it has been combined with the ornithine decarboxylase inhibitor DFMO in a large clinical trial for chemoprevention of sporadic colorectal adenocarcinoma [85, 86]. A striking chemopreventive effect was observed in the combination arm, with a 95 % decrease in advanced adenomas, which are most likely to progress to carcinoma [87]. Sulindac inhibition of COX resulted in inhibition of β-catenin signaling by enhancing its degradation in colon cancer [88]. Several studies have demonstrated the effectiveness of sulindac in reducing the size and number of adenomas in familial polyposis [77, 89]. Recently Stein et al. [90] reported that sulindac was a potent inhibitor of metastatic tumors in colon cancer [90]. There is a possibility that sulindac may exert an inhibitory effect on IFNγ-inducible chemokine expression and thus reduce the host immune response against tumorigenesis. Sulindac was shown to inhibit selectively IFNγ-induced expression of the chemokine CXCL9 in mouse macrophage RAW264.7 cells without affecting IFNγ-induced signal transducer and activator of transcription 1 (STAT1) activation [91]. The mechanism for the selective inhibition of CXCL9 is not yet known.
Many studies have shown that several PG synthesis inhibitors, such as indomethacin and piroxicam, suppress colon carcinogenesis in laboratory animal models [92–98]. Piroxicam was found to be an effective chemopreventive agent when administered in the diet during the initiation, post-initiation, and progression stages of colon carcinogenesis in laboratory rodents [92, 93]. Piroxicam at 200 and 400 ppm caused a dose-dependent decrease (45–64 %) in colon tumor incidence in F344 rats [93]. At a dose of 20 mg/day, it reduced mean rectal prostaglandin concentration by 50 % in individuals with a history of adenomas [99]. Bayer et al. [69] and Muller et al. [100] reported decreased expression of Treg-associated molecules such as forkhead box P3 (FOXp3) and IL-10 in indomethacin-treated tumors (Fig. 13.3). Based on evaluation of various immune cell markers [(HLA-DM, HLA-DO (peptide loading), HLA-DP, HLA-DQ, HLA-DR (antigen presentation), granzyme B, H, perforin and FCGR3A (CD16) (cytotoxicity), CD8+ cytotoxic T lymphocyte and CD4+ T helper cells]. Lonroth et al. [101] suggested that specific and nonspecific NSAIDs (indomethacin and celecoxib) alter colorectal cancer progression by affecting immune surveillance. They showed that indomethacin treatment increased in the infiltration of B cells, macrophages, CD4+ T helper cells, and CD8+ cytotoxic T lymphocyte and reduced Foxp3 expression in tumor tissue [101]. These NSAIDs have immunomodulatory effects in addition to their effects on AA metabolism that contribute to tumor inhibition.
Specific COX-2 inhibitors were designed to achieve greater efficacies and reduce GI toxicity. These drugs showed consistent strong chemopreventive effects in preclinical animal models [102, 103]. However, even the nonspecific COX inhibitor aspirin showed risk reductions similar in magnitude to specific COX-2 inhibitors. Studies by Talmadgea et al. [104] reported a significant expansion of immature myeloid suppressor cells (IMSCs) (phenotype Gr1+CD11b+ cells) and a reduction in CD4+ lymphocytes in the spleen during growth of 1,2-DMH-induced intestinal tumors in mice. The tumor-bearing mice showed increased expression of inflammatory molecules such as COX-2, iNOS, and arginine. Therapy with clinically relevant doses of celecoxib resulted in a reduced number of colon tumors and delayed tumor development by reducing immunosuppressive IMSCs. This study supports the role of COX-2 in inducing immunosuppressive activity during tumor development by causing apoptosis of CD4+ T cells. A population-based retrospective cohort study of individuals aged 65 years and older suggested that long-term use of non-aspirin NSAIDs was associated with reduced risk of CRC [105]. A randomized clinical trial with celecoxib showed significant reduction in the incidence of colonic adenomas [106, 107].
Chemopreventive effects of celecoxib and rofecoxib with daily standard dosages (200 and 25 mg, respectively) were demonstrated as a 69 % decrease in CRC. These drugs were approved in 1999; however, rofecoxib was withdrawn from the market in 2004 due to increased risk of cardiovascular events [108–110]. During this very short window of usage, both of these drugs showed significant reductions in CRC, but their associated risk of cardiotoxicity resulted in new searches for better COX inhibitors that retain potent preventive properties without the toxic effects. Significant inhibition of PanIN progression to PC in transgenic KrasG12D/+ mice upon dietary administration of the dual COX-LOX inhibitor licofelone has been reported by the authors [111] (Fig. 13.3). This study suggests the use/development of agents that can show well-balanced inhibition of COX and 5-LOX activities, with high efficacy in tumor inhibition and less or no toxicity. A complete blockade of progression of PanINs to PC was observed when licofelone was combined with gefitinib in transgenic KrasG12D/+ mice [112]. Hence, targeting multiple pathways with low nontoxic doses of potent agents is encouraging in prevention of PC.
Immunological effects of NSAIDs or non-NSAIDS in PC have not been studied much. The available literature on risk reduction in CRC and PC with NSAID usage suggests that chronic unresolved inflammation involving immune cells plays a critical role in tumor initiation, growth, and development. A thorough understanding of the functions of NKs, T cells, macrophages, and their related cytokines during different stages of tumor development is needed to help find effective approaches and to establish appropriate doses and administration schedules to suppress the tumors.
13.5 Immunomodulatory Effects of Statins
Since their discovery in 1976, statins have been in use for their beneficial effects in reducing serum cholesterol, low-density lipoprotein (LDL) and triglyceride levels and for increasing high-density lipoprotein (HDL) cholesterol [113]. Statins exhibit these effects by inhibiting HMG-CoA reductase in the cholesterol synthesis pathway (Fig. 13.4). Downstream metabolites of this pathway regulate posttranslational prenylation of Rho and Ras. Statins inhibit isoprenylation of Rho and Ras, which inhibits function of these proteins in cell mobility, shape, proliferation, differentiation, and survival. Since HDLs enhance anti-inflammatory responses by reducing cytokines (TNF-α, IL-1), statins also exhibit anti-inflammatory effects and are effective against inflammation and CRC in preclinical animal models. Although the beneficial effects of statins are predominantly via their lipid-lowering properties, they recently have been noted to have additional immunomodulatory actions that might exert beneficial effects. There is ample evidence that they exert anti-inflammatory and anti-oxidative actions and can induce tumor suppressors in colorectal cancer. A trial of statin therapy was conducted in Northern Israel between 1998 and 2004 in a population-based, case-controlled study of patients with a diagnosis of CRC. A modest reduction in CRC was shown in the general population without inflammatory bowel disease (IBD), but a substantial 94 % risk reduction was observed in a small number of IBD patients in a subset analysis [114]. A recent meta-analysis of 16 studies involving 1,692,863 participants and 7,807 PC cases showed no association between statin use and PC [115]. However, in a transgenic KrasG12D/+ mouse model, the authors and others have shown delayed progression of pancreatic lesions PanIN 1, PanIN 2, and PanIN 3 to PDAC in atorvastatin-treated mice compared with untreated controls via modulation of phosphatidylinositol 3-kinase (PI3K)/AKT signaling molecules [116]. Since the Akt pathway regulates Foxp3 expression and Treg development in the thymus, atorvastatin may have delayed PC development by decreasing Treg development [117, 118] (Fig. 13.4). Another study reported that fluvastatin and lovastatin reduced liver tumor formation and growth of established liver metastases in PC [119].
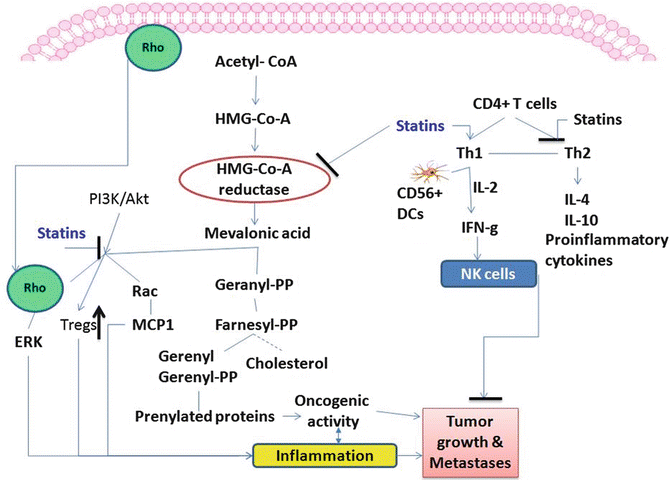
Fig. 13.4
Statins inhibit the cholesterol metabolite pathway and reduce the activation of Rho and Rac family proteins which are involved during tumor formation. Also, statins help in modulation of CD4+ T cells towards Th1 differentiation in the presence of CD56+ DCs that will activate NK cells which help in eliminating tumor cells. Further statins inhibit formation of pro-inflammatory cytokines
Although there are studies suggesting immunosuppressive function of statins, it was shown that statins act cooperatively with IL-2 to induce IFN-γ production in CD56dim NK cells [120–122] (Fig. 13.4). Previously, it was reported that statin effects on NK cells were inhibitory, but all of those studies focused on NK cell-target cell interactions with purified NK cells in co-culture with target cells [123–126]. In contrast, Gruenbacher et al. [121] reported that statins enhanced tumor death by increasing NK cell activity against tumor cells and increasing INF-γ production, effects mediated by CD56+HLA-DR+CD14+ DC-like accessory cells (Fig. 13.4). Atorvastatin and pravastatin showed anti-inflammatory properties by attenuating T-cell activation and proliferation [127, 128]. Mausner-Fainberg et al. [129] reported that treatment of human peripheral blood mononuclear cells (PBMCs) with atorvastatin, but not with mevastatin or pravastatin, increased the number of CD4+CD25high and CD4+CD25+Foxp3+ cells [129]. No difference was seen in Tregs in C57BL/6 mice with these agents. This report suggests that each statin is different in its mode of function. In trials of statin treatment for CRC, we showed decreased expression of COX-2 by rosuvastatin in AOM-induced colon tumors in rats, suggesting anti-inflammatory effects. Simvastatin was reported to reduce expression of the chemokines MCP1, MIP1α, and MIP1β as well as of the chemokine receptors CCR1, CCR2, CCR4, and CCR5 in human macrophages [130]. A clinical trial showed blocked expression of T-cell activation markers by atorvastatin [131]. A recent study showed that atorvastatin significantly decreased the expression of six cytokines (IL-6, IL-8, IL-1, plasminogen activator inhibitor type (PAI-1), TGF-β1, TGF-β) and five chemokines (CCL2, CCL7, CCL13, CCL18, CXCL1) and affected the expression of many inflammatory genes, as analyzed via DNA microarray analysis of human peripheral blood lymphocytes from normal subjects [132].
Fluvastatin demonstrated inhibitory activity against colitis and carcinogenesis in a mouse model, and it reduced oxidative DNA damage and activity of the DNA-synthesizing enzyme thymidine kinase in colorectal tissues [133]. Simvastatin was reported to cause significant reduction of tumor development by induction of apoptosis and suppression of angiogenesis in another colitis-associated CRC model. It also suppressed tumors by inhibiting VEGF and inducing apoptosis of tumor cells in a colon cancer xenograft model. Pitavastatin was effective in inhibiting AOM- and dextran sulfate sodium (DSS)-induced colitis-related colon carcinogenesis and significantly inhibited multiplicity of colon adenocarcinoma through modulation of mucosal inflammation, oxidative and nitrosative stress, and cell proliferation [134]. A 3-month treatment with 80 mg per day of atorvastatin resulted in reduced plasma chemokine CXCL10 levels and inflammation in Crohn’s disease patients [135]. Atorvastatin therapy was shown to reduce inflammation by reducing monocyte migration [136, 137]. The authors recently presented evidence for chemopreventive effects of rosuvastatin in AOM-induced colon tumors, with significant inhibition of the transformation from adenoma to adenocarcinoma [138]. Although this statin has the greatest lipid-lowering effect, concerns about its safety have been raised. It is evident from all these reports that considerable progress has been made in our understanding of some of the mechanisms underlying the beneficial effects of statins and that these include some actions besides inhibition of HMG-CoA reductase. The available preclinical animal data are supportive of chemopreventive properties of statins [133, 134, 138], but very little is known about the effects of these agents in high-risk subgroups, such as patients with resected colon cancer, and the effectiveness of statins as chemopreventive agents remains to be established in prospective randomized trials. Design and development of new statins and improved understanding of how statins manipulate immune cell functions is warranted to inhibit pro-inflammatory conditions in CRC and PC.
13.6 Immunomodulatory Effects of Selective Estrogen Receptor Modulators
17β-Estradiol is the most potent endogenous ligand for the estrogen receptor (ER)-β, but it binds equally well with ER-α and ER-β. The distribution and also the functions of these receptors are different in different tissues, and these features were behind the drive to identify or design selective ligands or selective estrogen receptor modulators (SERMS). Evidence gathered in recent years demonstrates roles for sex hormones in immunity. A 17β-estradiol- dependent increase in FOXp3 and PD1 expression with expansion of CD4+CD25+ T cells in mice has been suggested [139, 140]. And an enhanced functional activity of Tregs in suppressing immunity was observed upon 17β-estradiol treatment in mice [140]. It is evident that high Treg numbers and associated PD1 expression in CRC and PDAC are associated with poor prognosis. In ER-α knockout (KO) (Esr1–/–, ERKO) and ER-β KO (Esr2–/– (BERKO)) mice, Treg functional capacity and PD1 expression were significantly reduced compared with wild-type mice [140] (Fig. 13.5). Pretreatment with 17β-estradiol partially restored the functional Treg suppression of PD-1 KO mice [140].
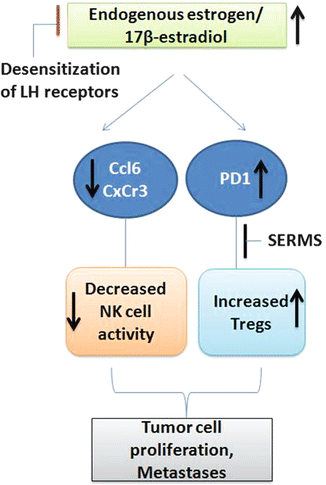
Fig. 13.5
Increased endogenous estrogen levels are linked to formation of more colonic tumors. They are reported to decrease the NK cell activating cytokines and chemokines and increase Tregs by increasing PD1 expression. Selective estrogen receptor modulators are reported to decrease formation of Tregs and CRC
17β-Estradiol is reported to enhance metastasis of both syngeneic and allogenic tumors in nude mice [141]. A link between female hormones and PC has been controversial. ERs were observed in both PC and normal pancreas [142], but a higher level of estrogen binding was reported in patient PC tissues compared with controls [142]. High ER-β RNA levels were reported in PC [143]. Specifically, ER-β2 mRNA was observed to play a more important role than ER-α in PC. However, a weak or no association of PC risk with the use of hormone replacement therapy (HRT) or contraceptives was reported in five cohort studies [144]. A similar result was reported in five case-controlled studies [144]. There is not much convincing data for a hormone dependence of PC, arguing against a role of estrogens or their antagonists in preclinical or clinical prevention or treatment of PC.
In nude mice with colon cancer xenografts, prolonged systemic administration of 17-estradiol suppressed NK activity resulting in frequent and large metastases to the liver and lungs and the mesenteric, omental, and mediastinal lymph nodes, supporting the role of NKs in controlling metastatic potential of primary tumors [145]. We have observed increased Ccl6 and Cxcr3, indicating increased NK cell activity, with suppressed colon tumor formation by decreasing endogenous estrogen production through desensitization of luteinizing hormone (LH) receptors in an ApcMin/+ mouse intestinal tumorigenesis model [146] (Fig. 13.5). Hence, the use of SERMs may provide protection in CRC by enhancing NK cell activity against tumor growth and suppressing Treg functions.
Anti-inflammatory properties of estrogens have been studied extensively [147]. Estrogen therapy lowers cytokine levels in postmenopausal women [148]. Estrogens modulate inflammatory genes through ER-α and ER-β. Since the discovery of ER-β, scientists have made enormous progress in understanding its biology. Its role is well established in the ovary, cardiovascular system, and brain, as well as in several animal models of inflammatory diseases, but less is known of its role in CRC and PC. Estrogens and their receptors are important regulators of colon physiology, but the roles in colonic epithelial cells are not well understood, and there is controversy about the role of ERs in inflammation and CRC [149]. One study reported that loss of ER-β did not cause any colonic tumor formation, suggesting that it is not associated with colonic neoplasia, nor were any significant differences found in colonic tumor formation in ApcMin/+ and ER-β−/− compound mice [150]. Some studies have indicated that ERs can suppress pro-inflammatory genes such as IL-6 and TNF-α [151–153], whereas both ER-α and ER-β have been implicated in repression of inflammatory genes by 17β-estradiol, with ER-β ligands exhibiting more potency in repressing genes induced by TNF-α [154].
Menopausal HRT use has been associated with a decreased colorectal cancer risk, suggesting that estrogen signaling is involved in colon physiology and cancer etiology. However, a French E3N prospective cohort study suggested that while menopausal HRT was not associated with significant colorectal adenoma or adenocarcinoma risk, any use of estrogens alone was associated with the risk of colorectal adenoma and cancer in opposite directions [155]. A very recent report suggests that estrogens promote tumor development in the context of inflammatory damage in a DSS-AOM mouse model. In this study, estrogen-treated animals had formation of invasive adenocarcinomas as compared with untreated animals. The pro-tumorigenic effect of estrogen was related to both ER-α and ER-β. A preclinical AOM-induced colitis model in an ER-β knockout mice revealed that ER-β-deficient animals had greater numbers and sizes of colon polyps with increased expression of IL-6, IL-17, and TNF-α. These results suggest that, in the presence of carcinogen insult, ER-β may provide protection against inflammatory conditions by healing mucosal damage. However, there is evidence suggesting a positive role of ER in enhancing colon tumor formation. The authors have reported an effect of the phytoestrogen genistein in enhancing AOM-induced colon carcinogenesis [156]. This result is consistent with evidence of the association of ER-β expression with elevated cell proliferation markers in tumors [157]. Data represented by the authors suggest that raloxifene, an ER-β antagonist, significantly inhibits AOM-induced formation total ACF (31–40 %) and multicrypt ACF (23–50 %) and suppresses colon adenocarcinoma multiplicity (to 3.28 ± 0.31 with 1.5 ppm raloxifene; to 2.96 ± 0.30 with 3 ppm raloxifene) in F344 rats [158, 159]. It is also observed >70 % fewer polyps with sizes of >1 mm upon raloxifene treatment in ApcMin/+ mice [16], and a significant decrease in AOM-induced colonic adenocarcinomas associated with inhibition of the Th2 cytokine IL-4 is found with raloxifene treatment [159] (Fig. 13.5).
Our findings and others suggest that modulation of immune cells by suppressing endogenous estrogen levels in CRC and targeting ERs may be useful as a potential preventive approach to modulate inflammatory responses in CRC [160]. Although the precise clinical indications still are being defined, the reported mechanistic and animal studies suggest that ER-modulating agents might represent a new class of drugs to prevent and treat inflammatory disorders. Ultimately, clinical trials with synthetic as well as natural plant-derived ER-β-selective compounds are needed to assess the potential translation of drugs that target ER-β for the prevention and treatment of human CRC associated with inflammatory disorders. No preclinical data exist on the use of SERMs in immunomodulation or prevention of PC.
13.7 Immunomodulatory Effects of Rexinoids
Retinoid X receptors (RXRs) and their rexinoid ligands regulate a family of genes having RXR receptor elements. They control differentiation, growth, and, in certain situations, induce apoptosis in tumor cells [161]. Rexinoids can cause cell differentiation and proliferation in T lymphocytes and inhibit activation and prevent apoptosis in B cells [162]. These effects are predominantly through the IL-2R in T and B cells; rexinoids may induce expression of IL-2Rα [163]. The RXR agonist AGN194204 enhanced Th2 development upon antigenic stimulation with splenic APCs [164] (Fig. 13.6). RXRα1/− mice were more susceptible to inflammation than are wild-type mice, and the RXR agonist LG101305 reduced IL-1β expression, providing genetic evidence that RXRs are protective and have anti-inflammatory functions [165]. A chemopreventive property of the rexinoid bexarotene in an ApcMin/+ colon cancer model is previously reported by the authors. It decreased the inflammatory cytokines granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-2 and IL-12, increased RXRα levels, and suppressed total intestinal polyps and colon tumors in male and female mice, by 38.2–9.9 % (P < 0.015 to P < 0.0001) or 8.5–36.9 %, respectively (P < 0.007) [166] (Fig. 13.6). A significant inhibition of colonic adenocarcinomas was observed with bexarotene alone and in combination with raloxifene treatment administered in the diet to F344 rats 8 weeks after AOM treatment [159]. Bexarotene-treated colonic tumors exhibited decreased IL-6 expression [159]. A recent study by Liby et al. [167] reported that the rexinoid LG100268 (LG268), alone or in combination with triterpenoids, increased survival in a LSL-KrasG12D /+;LSL-Trp53R172H/+;Pdx-1-Cre (KPC) triple mutant mouse model of PC. Collectively, these observations support a role for RXRs in immune cells, but, to our knowledge, there are no reports on the specific functions of RXRs in immune cells such as NKs, DCs, and Tregs in CRC and PC.
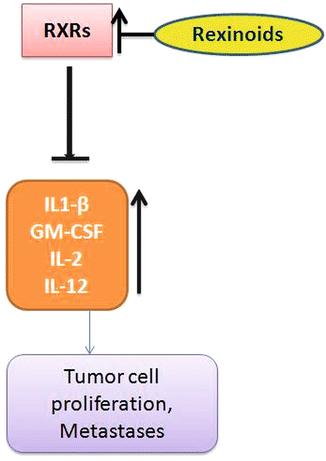
Fig. 13.6
Decreased expressions of RXRs are observed in CRC. Rexinoids increase RXR expression and decrease pro-inflammatory cytokines and increase the Th2 cytokine productions which will help in immunity development against tumor formation
13.8 Immunomodulatory Effects of Antidiabetic Agents
Many genes involved in diabetes play a role in cancer progression. There is mounting data suggesting that diabetics are more prone to certain cancers [168]. The antidiabetic drug metformin activates AMP kinase (pAMPK) in liver cells and inhibits production of glucose. Thus, it affects insulin signaling, whole body metabolism, and energy balance. Metformin was reported to increase immune memory cells and CD8+ T cells and to enhance tumor regression [169]. The use of metformin has been linked to a reduction in overall cancer incidence and specifically to reduction of breast cancer and PC. A report by Bodmer et al. [170] suggested that metformin was associated with a slightly increased risk of colon cancer (observed risk 1.43; 1.81 in men vs. 1.00 in women), and our results with metformin in CRC and PC are similar. In contrast, Hosono et al. [171] tested metformin for the inhibition of intestinal ACFs in BALB/c mice, where ACFs were formed after 6 weeks of AOM treatment and polyps after 32 weeks, and found a significant inhibitory effect on ACFs and a modest effect on polyps, suggesting that metformin may possess chemopreventive properties. The same group showed an inhibitory effect of metformin (250 mg/day) on preneoplastic ACF lesions in patients [172]. Another study in the Apc Min/+ mouse model observed that administration of metformin at 250 mg/kg in the diet significantly reduced the number of large polyps [173]. Metformin efficiently inhibited colon tumor growth in nude mice [174], and a significant decrease in PanINs and PDAC was observed in metformin-treated transgenic LSL-Kras mice [175, 176]. However, we observed a nonsignificant increase in AOM-induced CRC in metformin-treated F344 rats. Metformin-treated PC tissues had an increase in CD8+ T cells and a decrease in Tregs compared with control PC tissues. No effect of metformin on NK cell numbers or INFγ production in PC tissues was observed by the authors. Currently, a phase III clinical trial is recruiting patients for a study in which metformin plus modified FOLFOX 6 will be used in patients with metastatic PC. The primary outcome of this study will be to determine whether this treatment will have any effect on median survival (ClinicalTrials.gov ID-NCT01666730). The authors’ preclinical data in CRC suggest the need to evaluate metformin doses thoroughly and establish the mechanisms for its pro-tumorigenic effects in CRC and the conditions under which it may have any inhibitory effect in CRC. Studies of metformin in PC suggest that its tumor inhibitory action is based on pAMPK activation, reduced activity of mTOR, and immune-modulating effects. The potential inhibitory effects of metformin in PC support its further evaluation.
13.9 Immunomodulatory Effects of Natural Agents
The source of most biologically active compounds is plants. Not only are 25 % of all medicines derived from plants, but the starting material for many semisynthetic drugs is obtained from plant components. The compounds derived from plants are not only considered safer but are also cost-effective. Immunomodulatory functions of natural agents such as curcumin on T cells, DCs, and NKs have been investigated via cellular, immunological, biochemical, and molecular technologies and with the use of mouse models.
Curcumin has been shown to exhibit antitumor efficacy; however, clinical studies did not support adequate bioavailability with oral doses due to its poor absorption [177]. Effective delivery methods are under investigation to improve the efficacy of curcumin against CRC. High doses of curcumin (10 or 12 g/day) did result in a significant increase in serum levels in a clinical toxicology study [178], and some studies reported that 4 g/day of curcumin showed similar results [179]. In a double-blind study for ulcerative colitis, continuous consumption of 2 g/day curcumin with a standard diet provided significant protection against inflammation [180]. Fewer relapses were seen in the group taking curcumin. Similar effects were observed in ulcerative colitis and Crohn’s disease in another study [181]. These studies suggest that curcumin was able to reach the colonic tissue to exert its effects. Administration of 3.6 g/day of curcumin orally resulted in effective concentrations of 7.7 +/− 1.8 nmol/g in normal colorectal tissue and 12.7 +/− 5.7 umol/L in malignant tissue [182]. Curcumin was effective in modulating TNF-α, COX-2, and 5-LOX activities [183, 184]. Oral administration of curcumin (2–4 g per day for 30 days) in an open-label clinical trial in 44 eligible smokers with eight or more ACF on screening colonoscopy was found to reduce the number of ACFs significantly (40 %); however, a lower dose was ineffective [185]. This study suggests that, although systemic bioavailability is a problem, a significant inhibitory effect of curcumin at high concentrations was observed on ACF. Curcumin did not show any effect on proliferation markers or on AA metabolism. Immune modulatory effects of curcumin were not analyzed in this study.
A few reports on the immune-modulating effects of curcumin are available, but the mechanisms of action are not yet fully understood [186]. Available reports suggest that curcumin restores progenitor and effector circulating T cells. In an ApcMin/+ mouse model, curcumin injections led to an increase in intestinal CD4+ T cells and retarded growth of adenomas [187]. Varalakshmi et al. [188] reported that curcumin injections enhanced the mitogen- and antigen-induced proliferation potential of T cells, but did not impair or increase the cytotoxic potential of NK cells or Th1 regulatory cytokine production. However, it still is not clear whether curcumin functions similarly to provide stronger immune responses in the pathological conditions of CRC and PC. It has been previously reported that dietary administration of curcumin at different stages of colon tumor development had significant inhibitory effects on CRC. In CF1 mice, dietary administration of curcumin showed significant suppression of chemically induced forestomach, duodenal, and colon tumors when given during initiation and/or post-initiation periods of tumor development [189]. Curcumin also reduced early preneoplastic lesions, dysplasia, and ACF in the colons of rodents [189, 190]. In another study, administration of 0.8 and 1.6 % curcumin continuously during the initiation and post-initiation phases of tumor formation significantly reduced AOM-induced adenomas in rats [191]. A study by the authors showed similar results; continuous administration of 0.25 % curcumin during the initiation and post-initiation stages of tumor formation significantly inhibited both incidence and multiplicity of AOM-induced colon adenocarcinomas and the tumor burden in F344 rats [192]. Although preclinical efficacy data with curcumin have shown significant inhibitory effects in CRC, achieving clinically efficacious doses is still a challenge. Hence, combinations of curcumin with other natural agents including piperine or omega 3 fatty acids, or with agents that can enhance the uptake of curcumin to increase its bioavailability, may provide greater benefit against CRC.
Related posts:
 Immunopathology and Immunotherapy of Lymphoblastic Leukaemia
Immunotherapy for Pediatric Solid Tumors
Immunopathology of Specific Cancers in Males and Females and Immunotherapy of Prostate and Cervical Cancer
Immunology and Immunotherapy of Ovarian Cancer
Immunotherapy of Renal and Bladder Cancers
Immunology and Immunotherapy of Breast Cancer
Immunopathology and Immunotherapy of Lymphoblastic Leukaemia
Immunotherapy for Pediatric Solid Tumors
Immunopathology of Specific Cancers in Males and Females and Immunotherapy of Prostate and Cervical Cancer
Immunology and Immunotherapy of Ovarian Cancer
Immunotherapy of Renal and Bladder Cancers
Immunology and Immunotherapy of Breast Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


