Immune Landscape of Gastrointestinal Malignancy
Maggie Phillips
Gregory B. Lesinski
Introduction
Gastrointestinal (GI) tumors harbor a series of distinct immunologic features and are comprised of heterogeneous immune cell populations. The origins of many GI malignancies are closely related to inflammatory pathology, which may influence the landscape of cytokines, chemokines, and cellular constituents of these tumors. The failure of both endogenous and immunotherapy-induced antitumor immune responses across a range of GI malignancies is multifactorial. Several factors intrinsic to T cells themselves, such as inhibitory immune checkpoint receptors, or extrinsic to the T cells, including cytokines, chemokines, or suppressive cell types, comprise a redundant network that limits antitumor immunity. This dominant, suppressive immune phenotype can be characterized by canonical skewing of cytokine and chemokine profiles, paired with limited effector T-cell infiltration and a predominance of myeloid-lineage cells. Finally, the complexity of the microenvironment within GI tumors is also reflected in heterogeneous cancer-associated fibroblast (CAF) populations that facilitate cross-talk with both tumor and immune cells to regulate disease progression. This chapter provides an overview of key immunologic features of GI malignancies and provides a perspective related to priorities for future multidisciplinary research to advance the field.
The Microenvironment of GI Tumors Harbors Cellular Complexity
Malignancy can develop across multiple organ sites within the GI system, leading to aggressive tumors, many of which harbor key immunologic features. Complex cellular interactions with the immune system influence the course of disease and the response to immunotherapy (1). Thus, there is clinical importance in understanding the biology behind why these tumors are inherently nonresponsive to immunotherapy. Ultimately this could enable innovative treatment approaches for overcoming barriers to efficacy. One notable feature across GI cancer types is the striking cellular heterogeneity that exists within the tumor microenvironment (TME). It is now appreciated that a diverse array of cell populations can cooperate with transformed cells, further amplifying their aggressive behavior or shielding them from elimination by endogenous immune cells (Figure 11.1) (2). Key cellular components of the TME encompass vasculature, such as pericytes or endothelial cells,
the immune system, including lymphocytes or myeloid cells and fibroblast populations (3). Notably, CAFs play a major role in facilitating cross-talk and remodeling the architecture of the tumor and its surrounding stroma (4). This chapter will provide a brief overview on key immunologic features of GI tumors and our current understanding as to the role of various cellular components and soluble factors in this setting.
the immune system, including lymphocytes or myeloid cells and fibroblast populations (3). Notably, CAFs play a major role in facilitating cross-talk and remodeling the architecture of the tumor and its surrounding stroma (4). This chapter will provide a brief overview on key immunologic features of GI tumors and our current understanding as to the role of various cellular components and soluble factors in this setting.
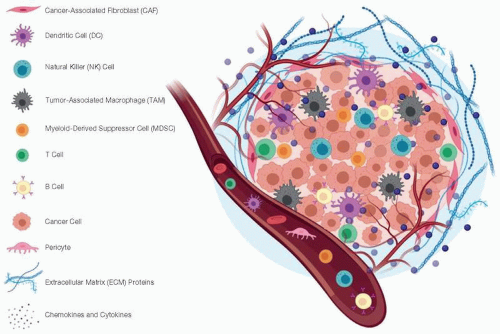 Figure 11.1 The tumor microenvironment (TME) of gastrointestinal (GI) cancers. The TME comprises complex interactions between both cellular and soluble components. Cellular cross-talk between immune cells, cancer-associated fibroblasts, and cancer cells can dictate disease progression and therapeutic response. These cellular components additionally secrete soluble factors, like chemokines and cytokines, that further influence the inflammatory TME of GI cancers. |
Inflammation Shapes the Immunologic Profile of GI Tumors
Given their anatomic origins, a salient feature present among most GI tumors is a close link with inflammatory pathology. There are many examples illustrating this relationship, including connections with viral infections and hepatocellular carcinoma, liver fluke infections with cholangiocarcinoma, Crohn’s- and colitis-associated colorectal cancer, and pancreatitis with pancreatic ductal adenocarcinoma (PDAC) (5,6,7,8). Furthermore, it is becoming increasingly appreciated that inflammation in the GI tract occurs concomitantly with alterations in the microbiome that may further impact cancer risk (9,10,11,12,13). Inflammation most certainly brings about dynamic change in GI tissues that are subject to oxidative damage, fibrosis, and an influx of lymphocytes, myeloid cells, and other innate immune cells that essentially render a failed attempt at restoring tissue homeostasis and repair (14). Together with underlying genomic aberrations in the epithelial cells residing in GI organs, this scenario perpetuates cellular transformation and tumor formation.
Cytokines and Chemokines Perpetuate the Inflammatory Nature of GI Tumors
The relationship between inflammation and GI malignancy clearly influences cytokine and immune cell profiles observed in these tumors. Dynamic interplay between cytokine or chemokine mediators is important for shaping the composition of immune cells within GI tumors. Many cytokines and chemokines are present at high levels either systemically in patients with GI tumors or within the TME. There is a high degree of redundancy in the functional consequences of this cytokine and chemokine dysregulation, which culminates in a suppressed antitumor
immune response. Some cytokines of notable importance in the setting of GI tumors that are supported by data from the literature include transforming growth factor-β (TGF-β), interleukin-6 (IL-6), IL-8, and vascular endothelial growth factor (VEGF), among others (15,16,17,18). In general, cytokine profiles are skewed away from the Th1 type, which can promote optimal immune responses from cytotoxic T and natural killer (NK) cells. Instead, Th2 and Th17 signatures are far more common in these patients, which enable maintenance of functionally suppressive immune cells including T regulatory cells (T regs), myeloid-derived suppressor cells (MDSC) or M2-polarized, tumor-associated macrophages (TAMs) (19,20,21).
immune response. Some cytokines of notable importance in the setting of GI tumors that are supported by data from the literature include transforming growth factor-β (TGF-β), interleukin-6 (IL-6), IL-8, and vascular endothelial growth factor (VEGF), among others (15,16,17,18). In general, cytokine profiles are skewed away from the Th1 type, which can promote optimal immune responses from cytotoxic T and natural killer (NK) cells. Instead, Th2 and Th17 signatures are far more common in these patients, which enable maintenance of functionally suppressive immune cells including T regulatory cells (T regs), myeloid-derived suppressor cells (MDSC) or M2-polarized, tumor-associated macrophages (TAMs) (19,20,21).
Other evidence supports a role for cytokines in the colony-stimulating factor family, such as granulocyte macrophage-colony-stimulating factor (GM-CSF), granulocyte-colony-stimulating factor (G-CSF), and macrophage-colony-stimulating factor (M-CSF), which enable preferential expansion of myeloid cells (22,23). Parallel to these immune changes, tumors confer other biologic advantages from cytokines and growth factors that enable angiogenesis including basic fibroblast growth factor (bFGF), IL-8, and VEGF. Using VEGF as an example, many of these factors can simultaneously influence multiple facets of the TME by concurrently enabling blood vessel formation and acting via its receptor that is expressed on immune cells to further promote expansion of T regs and MDSC. This cytokine profile can also be coupled with dysregulation of chemokine gradients in a manner that preferentially attracts suppressive immune cells. For example, GI tumors and their stroma can produce CXCL12/SDF-1 that limits access of T cells into the TME (24).
Data also continue to emerge linking distinct oncogenic driver mutations in tumor cells with production of specific chemokine mediators. One prominent example of this was from Li et al., who demonstrated oncogenic KRAS can enable myeloid infiltration in a manner that was dependent upon CXCL1 production by the tumor cells (25). Other connections between Wnt/β-catenin signaling or LKB1 mutations and reduced T-cell infiltration have also been noted (26,27). These observations support the idea that individualized immune signatures in the TME are subject to influence by the genomic features of the tumor itself.
Redundant Mechanisms of Immune Suppression Are Evident in GI Tumors
In general, many GI tumors are what is considered “immunologically cold,” meaning they lack abundant effector T-cell infiltrates. This is likely due to multiple factors, including but not limited to poor availability of neoantigens in tumors, inadequate chemokine gradients to enable their trafficking, or T-cell apoptosis upon entry into the hostile TME (20). Often, effector T cells are sequestered in regions distant from tumor cells, and those T cells that do gain access display phenotypic properties consistent with exhaustion. Other data indicate T-cell subsets including T regs or Th17 cells are present in these tumors, and may contribute to the impaired antitumor immune response. In contrast, myeloid cells and macrophages are major components within GI malignancies (28,29,30). These cells may be derived from tissue resident populations or alternatively are home to the microenvironment via the circulation. Taken together, the cellular composition of GI tumors enables multiple redundant mechanisms that inhibit productive antitumor immune responses. In general terms, mechanisms of limiting antitumor T-cell-mediated immune responses can be subdivided into two categories: (1) mechanisms that are intrinsic to the T cell itself (i.e., T-cell intrinsic) or (2) those mediated extrinsic soluble or cellular factors in the environment that feed back to limit T-cell function (i.e., T-cell extrinsic).
T-Cell Intrinsic Mechanisms Can Limit Antitumor Immune Responses
The most notable T-cell intrinsic mechanisms of suppressed antitumor immune responses are exemplified by inhibitory immune checkpoint receptors present on the surface on immune cell populations including T cells. Indeed, a wealth of knowledge is now available regarding the mechanism by which targeting the inhibitory programmed cell death protein 1 (PD-1) receptor and its interactions with ligands programmed death ligand-1 (PD-L1) and PD-L2 can perpetuate exhaustion phenotypes in T cells upon chronic antigen stimulation, thereby influencing antitumor immune response (reviewed in Pauken et al. [31]). The mechanism of
action of these therapies is likely far more complex as both PD-1 and its ligands are expressed on several different cell types (32). Likewise, other inhibitory immune checkpoints such as cytotoxic T- lymphocyte-associated protein-4 (CTLA-4) emerge more proximal to T-cell priming (33). Thus, the expression of PD-1, CTLA-4, and numerous other inhibitory receptors on T cells can enable functional impairment of antitumor immunity, and do so via distinct mechanisms (Figure 11.2). Importantly, multiple inhibitory immune checkpoint receptors have been characterized on the surface of T cells beyond PD-1, and these are often concurrently expressed, emphasizing their redundancy in function to sustain an immunologic advantage to tumors (34).
action of these therapies is likely far more complex as both PD-1 and its ligands are expressed on several different cell types (32). Likewise, other inhibitory immune checkpoints such as cytotoxic T- lymphocyte-associated protein-4 (CTLA-4) emerge more proximal to T-cell priming (33). Thus, the expression of PD-1, CTLA-4, and numerous other inhibitory receptors on T cells can enable functional impairment of antitumor immunity, and do so via distinct mechanisms (Figure 11.2). Importantly, multiple inhibitory immune checkpoint receptors have been characterized on the surface of T cells beyond PD-1, and these are often concurrently expressed, emphasizing their redundancy in function to sustain an immunologic advantage to tumors (34).
T-Cell Extrinsic Mechanisms Hinder Immunity against GI Malignancy
Complementing these hard-wired, T-cell-intrinsic suppressive mechanisms are a host of other cell types that limit antitumor immune responses via forces that are “extrinsic” to the T cells themselves. As discussed earlier, the landscape of immune cells populating GI tumors is dictated by cytokine and chemokine mediators. The net result is a limited number of both effector T cells, as well as cells with capacity to present tumor antigen and mount productive immune responses (20). One relevant example of this is recent evidence for IL-6-dependent apoptosis of type I dendritic cells (DC1) in pancreatic tumors (35). Importantly, this likely occurs early during the course of disease progression, even at the stage of PanIN lesions, further contributing to inadequate T-cell responses against these tumors. Myeloid cells including MDSC and TAM are prominent cellular components across the spectrum of GI tumors and suppress T-cell-mediated antitumor immune responses via several concurrent mechanisms. These include secretion of immune modulatory cytokines including TGF-β that directly limits cytotoxic capacity of T and NK cells, IL-10 that limits DC maturation and IL-6 that can promote MDSC expansion, and skewed T-cell profiles toward either T reg or Th17 phenotypes, depending on the other cytokines present (Figure 11.3) (36,37).
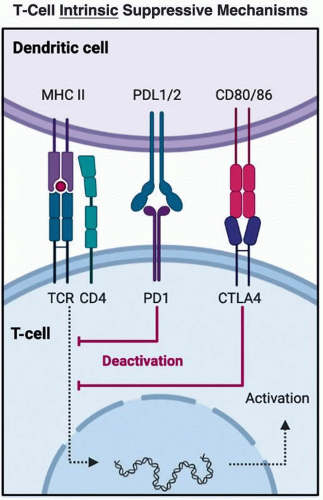 Figure 11.2 |
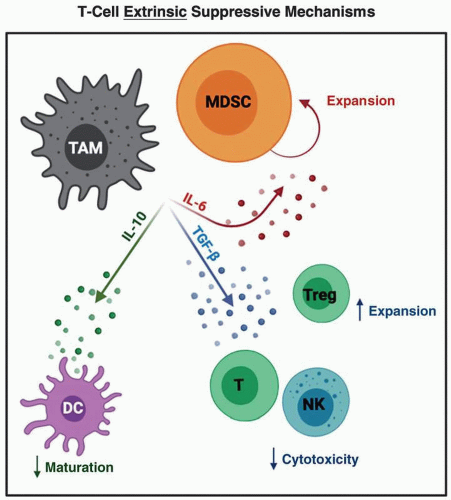 Figure 11.3 |
In a similar manner, B cells have been noted to secrete the cytokine IL-35 in several GI malignancies, resulting in exclusion of CD8+ T cells from the tumor (38,39). In addition to cytokine mediators, suppressive immune cells can serve as potent regulators of metabolic features within the TME. There are many examples of this (reviewed in Lyssiotis et al. [40]) including lactate, indoleamine-pyrrole 2,3-dioxygenase-mediated tryptophan catabolism, production of arginase and cysteine deprivation, among others (41). More recently, attention has turned to immune cells with more innate properties including gamma delta T cells or innate lymphoid cells (ILC2) as additional components of the suppressive TME (42,43). The sheer redundancy in cellular and soluble factors likely emphasizes the need for combination therapy approaches to derive the best chance at success in aggressive GI malignancies (44).
CAFs: At the Crossroads of Communication between Tumor and Immune Cells
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


